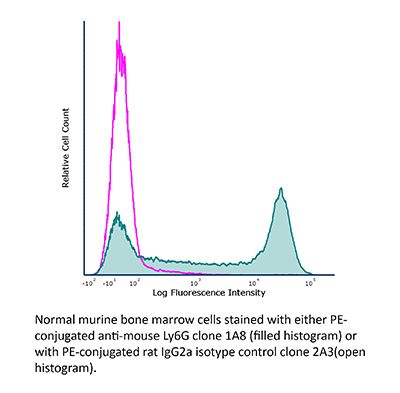
InVivoMAb rat IgG2a isotype control, anti-trinitrophenol
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107769 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
Bauche, D., et al. (2018). "LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis" Immunity 49(2): 342-352 e345.
PubMed
Interleukin-22 (IL-22)-producing group 3 innate lymphoid cells (ILC3) maintains gut homeostasis but can also promote inflammatory bowel disease (IBD). The regulation of ILC3-dependent colitis remains to be elucidated. Here we show that Foxp3(+) regulatory T cells (Treg cells) prevented ILC3-mediated colitis in an IL-10-independent manner. Treg cells inhibited IL-23 and IL-1beta production from intestinal-resident CX3CR1(+) macrophages but not CD103(+) dendritic cells. Moreover, Treg cells restrained ILC3 production of IL-22 through suppression of CX3CR1(+) macrophage production of IL-23 and IL-1beta. This suppression was contact dependent and was mediated by latent activation gene-3 (LAG-3)-an immune checkpoint receptor-expressed on Treg cells. Engagement of LAG-3 on MHC class II drove profound immunosuppression of CX3CR1(+) tissue-resident macrophages. Our study reveals that the health of the intestinal mucosa is maintained by an axis driven by Treg cells communication with resident macrophages that withhold inflammatory stimuli required for ILC3 function.
Kurtulus, S., et al. (2015). "TIGIT predominantly regulates the immune response via regulatory T cells" J Clin Invest. doi : 10.1172/JCI81187.
PubMed
Coinhibitory receptors are critical for the maintenance of immune homeostasis. Upregulation of these receptors on effector T cells terminates T cell responses, while their expression on Tregs promotes their suppressor function. Understanding the function of coinhibitory receptors in effector T cells and Tregs is crucial, as therapies that target coinhibitory receptors are currently at the forefront of treatment strategies for cancer and other chronic diseases. T cell Ig and ITIM domain (TIGIT) is a recently identified coinhibitory receptor that is found on the surface of a variety of lymphoid cells, and its role in immune regulation is just beginning to be elucidated. We examined TIGIT-mediated immune regulation in different murine cancer models and determined that TIGIT marks the most dysfunctional subset of CD8+ T cells in tumor tissue as well as tumor-tissue Tregs with a highly active and suppressive phenotype. We demonstrated that TIGIT signaling in Tregs directs their phenotype and that TIGIT primarily suppresses antitumor immunity via Tregs and not CD8+ T cells. Moreover, TIGIT+ Tregs upregulated expression of the coinhibitory receptor TIM-3 in tumor tissue, and TIM-3 and TIGIT synergized to suppress antitumor immune responses. Our findings provide mechanistic insight into how TIGIT regulates immune responses in chronic disease settings.
Ellis, G. T., et al. (2015). "TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection" EMBO Rep 16(9): 1203-1218.
PubMed
Streptococcus pneumoniae coinfection is a major cause of influenza-associated mortality; however, the mechanisms underlying pathogenesis or protection remain unclear. Using a clinically relevant mouse model, we identify immune-mediated damage early during coinfection as a new mechanism causing susceptibility. Coinfected CCR2(-/-) mice lacking monocytes and monocyte-derived cells control bacterial invasion better, show reduced epithelial damage and are overall more resistant than wild-type controls. In influenza-infected wild-type lungs, monocytes and monocyte-derived cells are the major cell populations expressing the apoptosis-inducing ligand TRAIL. Accordingly, anti-TRAIL treatment reduces bacterial load and protects against coinfection if administered during viral infection, but not following bacterial exposure. Post-influenza bacterial outgrowth induces a strong proinflammatory cytokine response and massive inflammatory cell infiltrate. Depletion of neutrophils or blockade of TNF-alpha facilitate bacterial outgrowth, leading to increased mortality, demonstrating that these factors aid bacterial control. We conclude that inflammatory monocytes recruited early, during the viral phase of coinfection, induce TRAIL-mediated lung damage, which facilitates bacterial invasion, while TNF-alpha and neutrophil responses help control subsequent bacterial outgrowth. We thus identify novel determinants of protection versus pathology in influenza-Streptococcus pneumoniae coinfection.
Dai, M., et al. (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138.
PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
Ngiow, S. F., et al. (2015). "A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1" Cancer Res 75(18): 3800-3811.
PubMed
Despite successes, thus far, a significant proportion of the patients treated with anti-PD1 antibodies have failed to respond. We use mouse tumor models of anti-PD1 sensitivity and resistance and flow cytometry to assess tumor-infiltrating immune cells immediately after therapy. We demonstrate that the expression levels of T-cell PD1 (PD1(lo)), myeloid, and T-cell PDL1 (PDL1(hi)) in the tumor microenvironment inversely correlate and dictate the efficacy of anti-PD1 mAb and function of intratumor CD8(+) T cells. In sensitive tumors, we reveal a threshold for PD1 downregulation on tumor-infiltrating CD8(+) T cells below which the release of adaptive immune resistance is achieved. In contrast, PD1(hi) T cells in resistant tumors fail to be rescued by anti-PD1 therapy and remain dysfunctional unless intratumor PDL1(lo) immune cells are targeted. Intratumor Tregs are partly responsible for the development of anti-PD1-resistant tumors and PD1(hi) CD8(+) T cells. Our analyses provide a framework to interrogate intratumor CD8(+) T-cell PD1 and immune PDL1 levels and response in human cancer. Cancer Res; 75(18); 3800-11. (c)2015 AACR.
Xiao, N., et al. (2014). "The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells" Nat Immunol 15(7): 657-666.
PubMed
Follicular helper T cells (T(FH) cells) are responsible for effective B cell-mediated immunity, and Bcl-6 is a central factor for the differentiation of T(FH) cells. However, the molecular mechanisms that regulate the induction of T(FH) cells remain unclear. Here we found that the E3 ubiquitin ligase Itch was essential for the differentiation of T(FH) cells, germinal center responses and immunoglobulin G (IgG) responses to acute viral infection. Itch acted intrinsically in CD4(+) T cells at early stages of T(FH) cell development. Itch seemed to act upstream of Bcl-6 expression, as Bcl-6 expression was substantially impaired in Itch(-/-) cells, and the differentiation of Itch(-/-) T cells into T(FH) cells was restored by enforced expression of Bcl-6. Itch associated with the transcription factor Foxo1 and promoted its ubiquitination and degradation. The defective T(FH) differentiation of Itch(-/-) T cells was rectified by deletion of Foxo1. Thus, our results indicate that Itch acts as an essential positive regulator in the differentiation of T(FH) cells.
Walsh, K. B., et al. (2014). "Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy" J Virol 88(11): 6281-6293.
PubMed
The cytokine storm is an intensified, dysregulated, tissue-injurious inflammatory response driven by cytokine and immune cell components. The cytokine storm during influenza virus infection, whereby the amplified innate immune response is primarily responsible for pulmonary damage, has been well characterized. Now we describe a novel event where virus-specific T cells induce a cytokine storm. The paramyxovirus pneumonia virus of mice (PVM) is a model of human respiratory syncytial virus (hRSV). Unexpectedly, when C57BL/6 mice were infected with PVM, the innate inflammatory response was undetectable until day 5 postinfection, at which time CD8(+) T cells infiltrated into the lung, initiating a cytokine storm by their production of gamma interferon (IFN-gamma) and tumor necrosis factor alpha (TNF-alpha). Administration of an immunomodulatory sphingosine-1-phosphate (S1P) receptor 1 (S1P1R) agonist significantly inhibited PVM-elicited cytokine storm by blunting the PVM-specific CD8(+) T cell response, resulting in diminished pulmonary disease and enhanced survival. IMPORTANCE: A dysregulated overly exuberant immune response, termed a “cytokine storm,” accompanies virus-induced acute respiratory diseases (VARV), is primarily responsible for the accompanying high morbidity and mortality, and can be controlled therapeutically in influenza virus infection of mice and ferrets by administration of sphingosine-1-phosphate 1 receptor (S1P1R) agonists. Here, two novel findings are recorded. First, in contrast to influenza infection, where the cytokine storm is initiated early by the innate immune system, for pneumonia virus of mice (PVM), a model of RSV, the cytokine storm is initiated late in infection by the adaptive immune response: specifically, by virus-specific CD8 T cells via their release of IFN-gamma and TNF-alpha. Blockading these cytokines with neutralizing antibodies blunts the cytokine storm and protects the host. Second, PVM infection is controlled by administration of an S1P1R agonist.
Mittal, D., et al. (2014). "Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor" Cancer Res 74(14): 3652-3658.
PubMed
Adenosine targeting is an attractive new approach to cancer treatment, but no clinical study has yet examined adenosine inhibition in oncology despite the safe clinical profile of adenosine A2A receptor inhibitors (A2ARi) in Parkinson disease. Metastasis is the main cause of cancer-related deaths worldwide, and therefore we have studied experimental and spontaneous mouse models of melanoma and breast cancer metastasis to demonstrate the efficacy and mechanism of a combination of A2ARi in combination with anti-PD-1 monoclonal antibody (mAb). This combination significantly reduces metastatic burden and prolongs the life of mice compared with either monotherapy alone. Importantly, the combination was only effective when the tumor expressed high levels of CD73, suggesting a tumor biomarker that at a minimum could be used to stratify patients that might receive this combination. The mechanism of the combination therapy was critically dependent on NK cells and IFNgamma, and to a lesser extent, CD8(+) T cells and the effector molecule, perforin. Overall, these results provide a strong rationale to use A2ARi with anti-PD-1 mAb for the treatment of minimal residual and metastatic disease.
Simons, D. M., et al. (2013). "Autoreactive Th1 cells activate monocytes to support regional Th17 responses in inflammatory arthritis" J Immunol 190(7): 3134-3141.
PubMed
We have examined mechanisms underlying the formation of pathologic Th17 cells using a transgenic mouse model in which autoreactive CD4(+) T cells recognize influenza virus hemagglutinin (HA) as a ubiquitously expressed self-Ag and induce inflammatory arthritis. The lymph nodes of arthritic mice contain elevated numbers of inflammatory monocytes (iMO) with an enhanced capacity to promote CD4(+) Th17 cell differentiation, and a regional inflammatory response develops in the paw-draining lymph nodes by an IL-17-dependent mechanism. The activation of these Th17-trophic iMO precedes arthritis development and occurs in the context of an autoreactive CD4(+) Th1 cell response. Adoptive transfer of HA-specific CD4(+) T cells into nonarthritic mice expressing HA as a self-Ag similarly led to the formation of Th1 cells and of iMO that could support Th17 cell formation, and, notably, the accumulation of these iMO in the lymph nodes was blocked by IFN-gamma neutralization. These studies show that autoreactive CD4(+) Th1 cells directed to a systemically distributed self-Ag can promote the development of a regional Th17 cell inflammatory response by driving the recruitment of Th17-trophic iMO to the lymph nodes.
Bamboat, Z. M., et al. (2010). "Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury" Hepatology 51(2): 621-632.
PubMed
Endogenous ligands such as high-mobility group box 1 (HMGB1) and nucleic acids are released by dying cells and bind Toll-like receptors (TLRs). Because TLR9 sits at the interface of microbial and sterile inflammation by detecting both bacterial and endogenous DNA, we investigated its role in a model of segmental liver ischemia-reperfusion (I/R) injury. Mice were subjected to 1 hour of ischemia and 12 hours of reperfusion before assessment of liver injury, cytokines, and reactive oxygen species (ROS). Wild-type (WT) mice treated with an inhibitory cytosine-guanosine dinucleotide (iCpG) sequence and TLR9(-/-) mice had markedly reduced serum alanine aminotransferase (ALT) and inflammatory cytokines after liver I/R. Liver damage was mediated by bone marrow-derived cells because WT mice transplanted with TLR9(-/-) bone marrow were protected from hepatic I/R injury. Injury in WT mice partly depended on TLR9 signaling in neutrophils, which enhanced production of ROS, interleukin-6 (IL-6), and tumor necrosis factor (TNF). In vitro, DNA released from necrotic hepatocytes increased liver nonparenchymal cell (NPC) and neutrophil cytokine secretion through a TLR9-dependent mechanism. Inhibition of both TLR9 and HMGB1 caused maximal inflammatory cytokine suppression in neutrophil cultures and conferred even greater protection from I/R injury in vivo. CONCLUSION: TLR9 serves as an endogenous sensor of tissue necrosis that exacerbates the innate immune response during liver I/R. Combined blockade of TLR9 and HMGB1 represents a clinically relevant, novel approach to limiting I/R injury.
Bamboat, Z. M., et al. (2010). "Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion" J Clin Invest 120(2): 559-569.
PubMed
TLRs are recognized as promoters of tissue damage, even in the absence of pathogens. TLR binding to damage-associated molecular patterns (DAMPs) released by injured host cells unleashes an inflammatory cascade that amplifies tissue destruction. However, whether TLRs possess the reciprocal ability to curtail the extent of sterile inflammation is uncertain. Here, we investigated this possibility in mice by studying the role of conventional DCs (cDCs) in liver ischemia/reperfusion (I/R) injury, a model of sterile inflammation. Targeted depletion of mouse cDCs increased liver injury after I/R, as assessed by serum alanine aminotransferase and histologic analysis. In vitro, we identified hepatocyte DNA as an endogenous ligand to TLR9 that promoted cDCs to secrete IL-10. In vivo, cDC production of IL-10 required TLR9 and reduced liver injury. In addition, we found that inflammatory monocytes recruited to the liver via chemokine receptor 2 were downstream targets of cDC IL-10. IL-10 from cDCs reduced production of TNF, IL-6, and ROS by inflammatory monocytes. Our results implicate inflammatory monocytes as mediators of liver I/R injury and reveal that cDCs respond to DAMPS during sterile inflammation, providing the host with protection from progressive tissue damage.
Product Citations
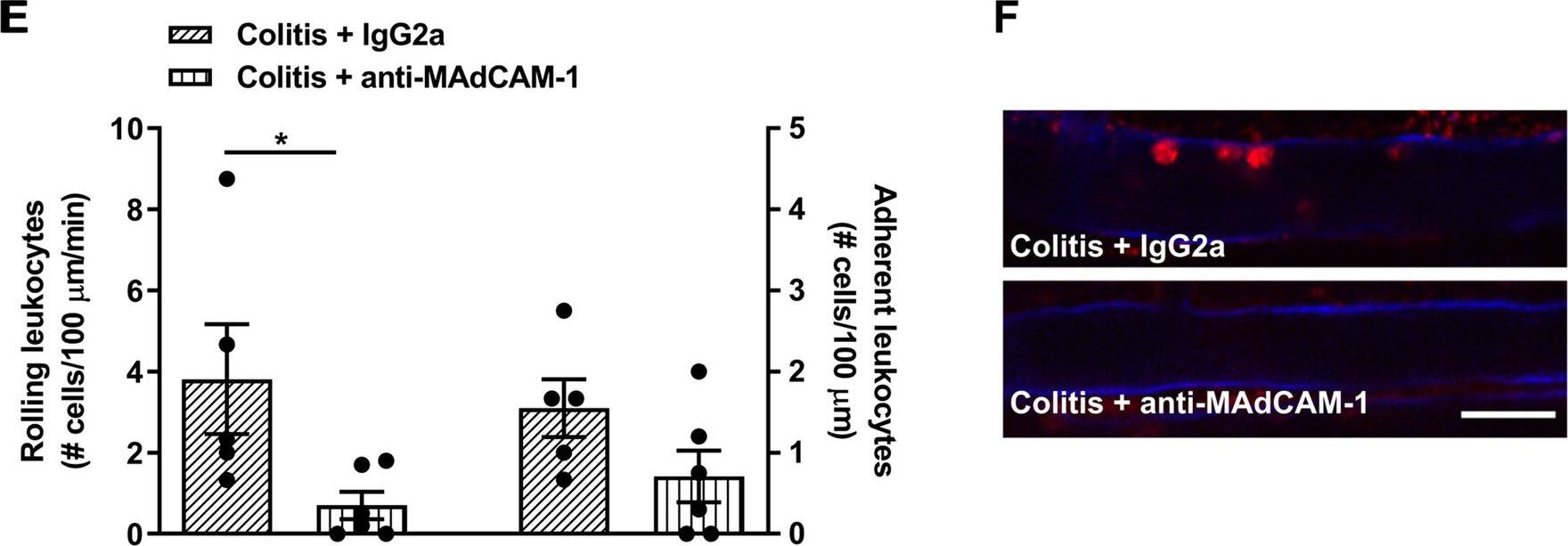
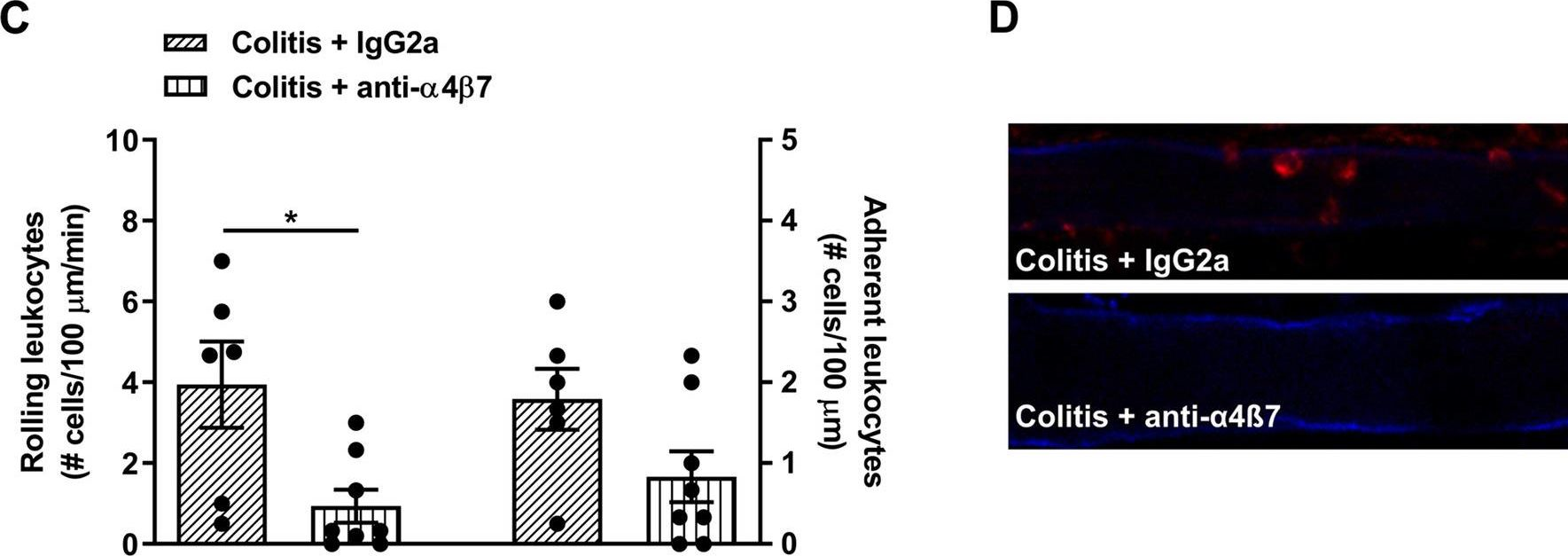
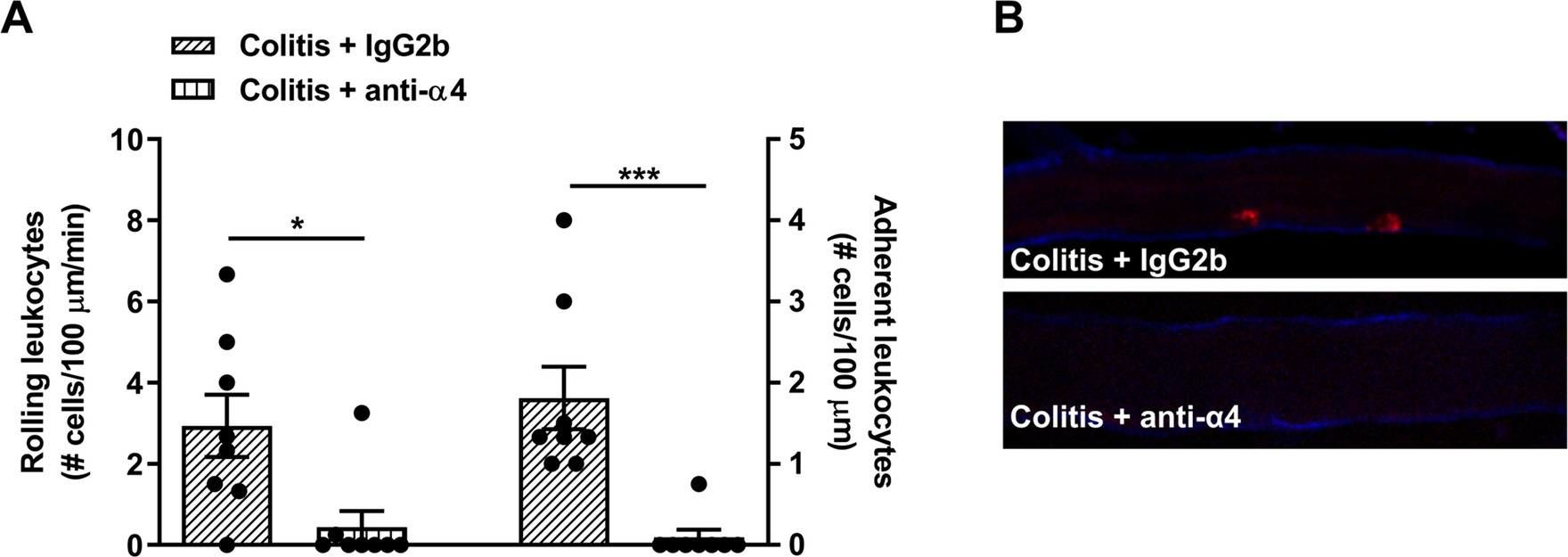
-
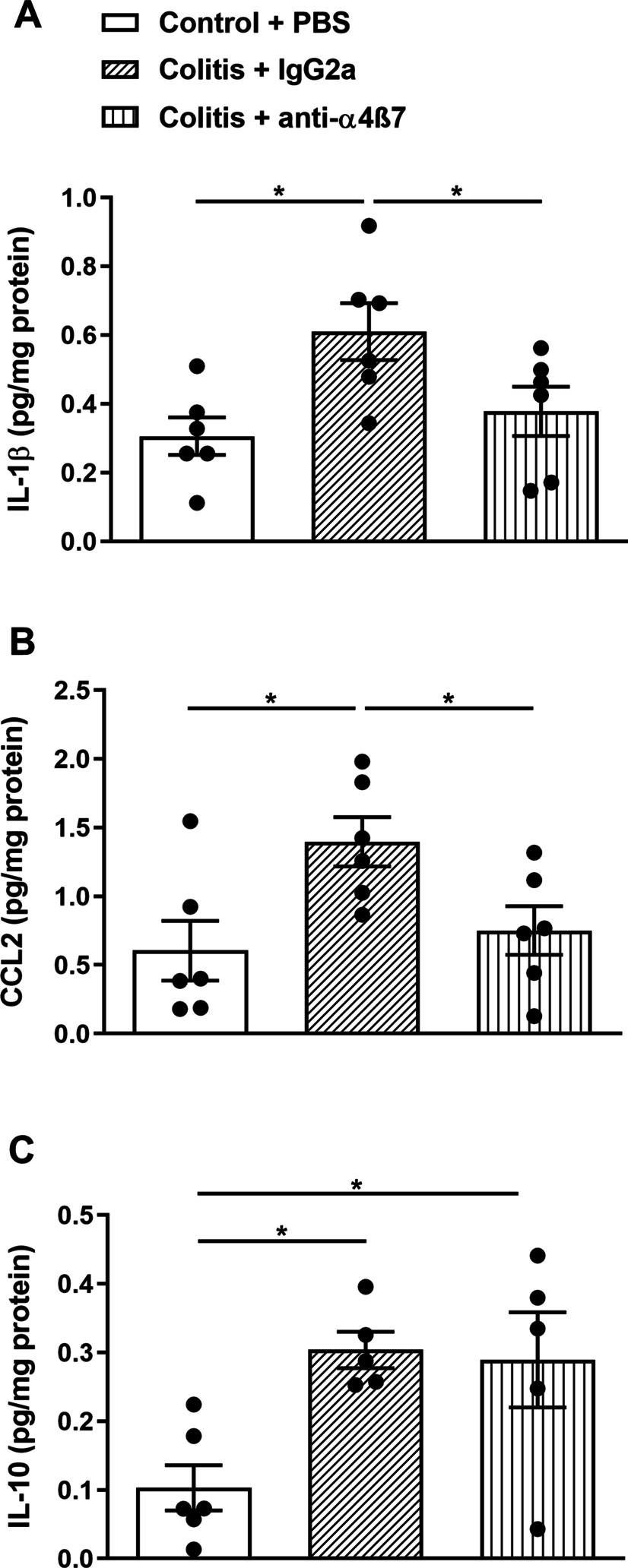
AhR Activation Transcriptionally Induces Anti-Microbial Peptide Alpha-Defensin 1 Leading to Reversal of Gut Microbiota Dysbiosis and Colitis.
In Gut Microbes on 1 December 2025 by Palrasu, M., Kakar, K., et al.
PubMed
Alpha-defensin 1 is a small antimicrobial peptide that acts as the first line of defense against pathogens. It is induced following microbial cues and inflammatory signals in neutrophils and Paneth cells in the small intestine, which suggests that it plays a role in microbial homeostasis in the gut. The gut microbial products also serve as ligands for the aryl hydrocarbon receptor (AhR), an environmental sensor. In the current study, we investigated if there is any crosstalk between AhR and alpha-defensin 1. Interestingly, we found a positive correlation between AhR and alpha-defensin 1 protein levels in ileal tissues from active Crohn's' (CD) patients and epithelial cells (IECs) from multiple models of murine colitis. In vitro downregulation of AhR led to inhibition of α-defensin 1, while activation of AhR induced α-defensin 1 in IECs. AhR directly targeted the dioxin response element 3 (DRE3) region on the α-defensin 1 promoter in IECs. AhR-mediated induction of α-defensin 1 in colitis mice reversed the gut microbial dysbiosis and alleviated colitis. Our data identify a novel signaling pathway in which AhR acts as a transcription factor for α-defensin 1, leading to regulation of homeostasis between gut microbiota, intestinal mucosa, and mucosal immunity.
-
-
Cancer Research
-
Immunology and Microbiology
Targeting the atypical chemokine receptor 2 (Ackr2) improves the benefit of anti-PD-1 immunotherapy in melanoma mouse model.
In Oncoimmunology on 1 December 2025 by Noman, M. Z., Szpakowska, M., et al.
PubMed
Immune checkpoint blockade (ICB) therapies, such as anti-PD-1, have transformed cancer treatment, but many patients do not respond due to a non-inflammatory tumor microenvironment (TME). Here, we investigated the impact of targeting Atypical Chemokine Receptor 2 (ACKR2), which scavenges key chemokines involved in immune cell recruitment, on the improvement of anti-PD-1-based therapy. In a melanoma mouse model, we demonstrated that Ackr2 inhibition increases the release of proinflammatory chemokines CCL5 and CXCL10 and enhances the infiltration of NK cells, activated CD8+ and CD4+ effector T cells while reducing regulatory T cells (Tregs) in the TME. Targeting Ackr2 led to tumor growth inhibition, improved survival, and enhanced response to anti-PD-1 therapy. In BRAF- and NRAS-mutant melanoma patients, low ACKR2 expression or high CCL5/CXCL10 levels correlated with improved survival and higher CD8+ T cell markers. Targeting ACKR2 represents a promising approach for developing combination therapies, particularly for 'cold' ICB resistant tumors.
-
-
-
Cancer Research
Metabolomic and transcriptomic profiling of HNSCC identifies AMIGO2 as a therapeutic target modulating tumor microenvironment.
In NPJ Precis Oncol on 18 November 2025 by Liu, G., Yao, X., et al.
PubMed
Extensive studies have demonstrated the relationship between metabolic reprogramming and the tumor microenvironment. Here, we characterized the head and neck squamous cell carcinoma (HNSCC) evolutionary landscape using spatial metabolomics/transcriptomics, single-cell transcriptomics, and bulk multi-omics. Metabolic heterogeneity during HNSCC malignant transformation was identified, with significant enrichment in the purine metabolism. Integrating single-cell and bulk data, we developed a robust ligand-receptor-based signature (LRS) linked to NT5E, a key upstream regulator of purine metabolism, which served as an independent prognostic indicator. The low LRS subtype was associated with a high proportion of immune cell infiltration and improved response to immunotherapy. Notably, in vitro and in vivo experiments demonstrated that AMIGO2, a core molecule within the LRS, regulates tumor-associated purine metabolism, and that its downregulation suppresses tumor cell invasion and migration, inhibits myofibroblast differentiation, and promotes immune effector cell infiltration. Moreover, combining AMIGO2 targeting with anti-PD-1 therapy yielded superior efficacy. Consistent validation was also obtained in a clinical cohort of HNSCC and premalignancy patients.
-
-
-
Cancer Research
-
Immunology and Microbiology
TAMs-mediated resistance to oncolytic virus M1 in solid tumors.
In J Immunother Cancer on 13 November 2025 by Liang, X., Li, J., et al.
PubMed
Oncolytic virus M1 (OVM), a naturally occurring alphavirus, has demonstrated potent antitumor activity in various solid tumor models by inducing immunogenic cell death and activating CD8+ T cells. However, its in vivo efficacy varies widely, and resistance mechanisms remain poorly understood. Tumor-associated macrophages (TAMs), key immunosuppressive cells within the tumor microenvironment, may limit OVM therapeutic potential.
-
-
Mucoricin binding to β-glucan sites on germinating Mucorales spores disrupts neutrophil swarming to promote pathogenicity
In bioRxiv on 29 October 2025 by Baimpa, S., Sertedakis, M., et al.
-
-
Cancer Research
-
Immunology and Microbiology
VEGFR2 blockade converts thermally ablative focused ultrasound into a potent driver of T cell-dependent anti-tumor immunity
In bioRxiv on 24 October 2025 by Schwartz, M. R., Anwar, N. Z., et al.
-
-
-
Cancer Research
-
Immunology and Microbiology
OCA-B/Pou2af1 Expression in Mouse T Cells Promotes PD-1 Blockade-Induced Autoimmunity but is Dispensable for Anti-Tumor Immunity
In bioRxiv on 23 October 2025 by Du, J., Manna, A. K., et al.
-
-
-
Immunology and Microbiology
-
Genetics
-
Cancer Research
Epigenetic suppression of Nrf2-Slc40a1 axis induces ferroptosis and enhances immunotherapy in pancreatic cancer.
In J Immunother Cancer on 23 October 2025 by Zhang, Y., Yu, R., et al.
PubMed
Despite progress in immunotherapy for several solid tumors, pancreatic ductal adenocarcinoma (PDAC) remains largely unresponsive, primarily due to its profoundly immunosuppressive tumor microenvironment (TME) characterized by limited CD8+ T cell infiltration. Novel strategies are needed to overcome this immune resistance and enhance the efficacy of checkpoint blockade.
-
-
-
Cancer Research
-
Immunology and Microbiology
Tumor-intrinsic MHC-II activation in pancreatic ductal adenocarcinoma enhances immune response and treatment efficacy
In bioRxiv on 22 October 2025 by Chen, C., Gribbin, K. P., et al.
-
-
-
Cancer Research
-
Immunology and Microbiology
Enhancing melanoma treatment through systemic delivery of an immune boosting Staphylococcus epidermidis strain.
In Sci Rep on 21 October 2025 by Hwang, J., Kim, G., et al.
PubMed
A unique strain of Staphylococcus epidermidis, AIT01 (AIT, Airway Immune Trainer), identified in our previous research, has demonstrated immune-boosting properties. This study aimed to evaluate the systemic immune-modulatory effects and potential anti-tumor properties of this immune-enhancing skin microbiota strain. A series of ex vivo and in vivo experiments were conducted to assess immune cell proliferation, cytokine production, and anti-tumor efficacy. In ex vivo studies, splenocytes treated with the bacterial lysate or culture supernatant of the strain showed significantly increased viability in a concentration-dependent manner. Flow cytometry analysis revealed increased populations of dendritic cells, NK cells (Natural killer cells), and γδ T cells, with enhanced cytokine production, particularly IFN-γ (Interferon-γ) and perforin, in the lysate-treated group. When administered via intraperitoneal and intravenous routes in vivo, mice showed significant inhibition of melanoma growth upon receiving the bacterial lysate. Notably, pre-treatment demonstrated superior efficacy compared to post-treatment. Furthermore, the combination of the bacterial lysate with anti-PD-1 (anti-Programmed cell death protein-1) monoclonal antibody further suppressed tumor growth compared to anti-PD-1 monotherapy. These findings suggest that the AIT01 lysate enhances immune cell proliferation and cytokine production, contributing to its potent anti-tumor effects. The systemic delivery of this immune-boosting skin microbiota strain, particularly in combination with anti-PD-1 therapy, holds promise as an effective immunotherapeutic strategy against melanoma.
-
-
-
Immunology and Microbiology
EP300 compromises antitumor immunity by increasing SOCS1 expression.
In J Immunother Cancer on 15 October 2025 by Zeng, Y., Zhou, Y., et al.
PubMed
Beyond supporting cancer cell proliferation, tumor growth relies on the ability of cancer cells to evade immune surveillance. Identifying novel molecules that promote tumor immune escape may help develop more effective immunotherapeutic strategies. The histone acetyltransferase E1A-binding protein p300 (EP300) is a key epigenetic regulator that modulates gene transcription through chromatin remodeling and acetylation of histones and transcription factors. However, its role in regulating immune evasion remains incompletely understood. This study investigates the impact of EP300 on tumor immune escape and suggests its potential as an immunotherapeutic target.
-
-
-
Cancer Research
-
Immunology and Microbiology
TNG260 Is a Small-Molecule CoREST Inhibitor That Sensitizes STK11-Mutant Tumors to Anti-PD-1 Immunotherapy.
In Cancer Res on 15 October 2025 by Ahronian, L. G., Sahu, S., et al.
PubMed
Patients with non-small cell lung cancer (NSCLC) with loss of the tumor suppressor gene STK11 are resistant to immune checkpoint therapies like anti-PD-1. In this study, we conducted an in vivo CRISPR screen that identified histone deacetylase 1 as a target to reverse anti-PD-1 resistance driven by loss of STK11 and developed TNG260, a potent small-molecule inhibitor of the CoREST complex with selectivity exceeding previously generated inhibitors in this class in preclinical studies. Treatment with TNG260 led to increased expression of immunomodulatory genes in STK11-deficient cancer cells. When combined with anti-PD-1, TNG260 induced immune-mediated stasis and/or regression in STK11-deficient syngeneic tumor models and autochthonous NSCLC models. In the tumors of patients with STK11-deficient cancers in a clinical trial (NCT05887492), treatment with a combination of TNG260 and pembrolizumab increased intratumoral histone acetylation, PD-L1 tumor proportion scores, and T-cell infiltration into the tumor microenvironment. This study illustrates a promising treatment strategy for addressing immune evasion in patients with STK11-mutant NSCLC.
-
-
-
COVID-19
The hyaluronan receptor CD44 drives COVID-19 severity through its regulation of neutrophil migration
In bioRxiv on 14 October 2025 by Hart, D. J., Uddin, M. J., et al.
-
-
Hydroxyapatite microspheres induce durable pleurodesis and are rapidly cleared by pleural osteoclasts.
In JCI Insight on 8 October 2025 by Tanaka, Y., Takahashi, Y., et al.
PubMed
Talc pleurodesis is highly effective for preventing recurrence of pneumothorax and pleural effusion, but it can be complicated by dissemination, acute lung injury, lead exposure, and foreign body-induced chronic inflammation and pain. Our objective is to develop a safe, biodegradable, contaminant-free particle for pleurodesis. We used mouse models of pneumothorax and malignant pleural effusion to compare the efficacy and safety of pleurodesis with talc and hydroxyapatite microspheres (HAM). Intrapleural instillation of microspheres induced pleural adhesions, fibrosis, and symphysis as effectively as talc and resulted in more durable protection from experimental pneumothorax. HAM and talc both induced an osteoclastogenic, inflammatory, and fibrotic response in pleural lavage cells. Intrapleural HAM was resorbed by osteoclast action over 3 months, whereas talc was not cleared. Deletion of the osteoclast effector, CTSK, diminished pleural adhesion formation and fibrosis by HAM, and inhibition of osteoclastogenesis with anti-RANKL antibody delayed HAM clearance. We found no difference in activity level, feeding behavior, or lung compliance between particles, but talc induced more persistent pleural inflammation. We conclude that HAM resulted in an osteoclastogenic and fibrogenic pleural response that induced pleurodesis that was more durable than talc with a superior safety profile due in part to osteoclast-mediated particle clearance.
-
-
Cancer Research
Preclinical models of melanoma leptomeningeal disease to assess intrathecal checkpoint blockade.
In Sci Rep on 2 October 2025 by Guerrieri, R. A., Fischer, G. M., et al.
PubMed
Leptomeningeal disease (LMD) is a subtype of central nervous system metastatic disease that is associated with poor patient outcomes and limited treatment options. There is an unmet need to develop preclinical models of LMD to expedite and improve the development of new therapeutics. Here, we describe the development of multiple orthotopic immunocompetent murine models of melanoma LMD, including their use to assess the efficacy of systemic and/or intrathecal immunotherapy. LMD was established by direct intrathecal injection of murine cell lines (B16-F10, BP, D4M, D4M-UV2, MC38-gp100, RMS, YUMM3.1, and YUMMER1.7) into the cisterna magna of C57BL/6 mice. Tumor take rate, distribution, histology, peri-procedural mortality, and animal survival were assessed for each cell line. Intrathecal and systemic treatment with anti-PD1 were tested for safety, efficacy, and immune infiltration for LMD. Cisternal injection of murine melanoma cell lines successfully established LMD with low peri-procedural mortality, high tumor take rate, and varied survival duration across the panel of cell lines. Decreasing the total number of cells injected and increasing the volume of suspension of the injected cells increased the rate of distal spinal cord deposits, reflecting the common clinical distribution of LMD. Intrathecal administration of anti-PD1 in non-tumor bearing mice caused no morbidity or toxicity. Concurrent intrathecal and systemic anti-PD1 immunotherapy increased the survival of mice with murine melanoma LMD. We have established and characterized several immunocompetent murine models of LMD to facilitate the development and testing of new, more effective immunotherapy strategies for melanoma patients with LMD.
-
-
-
Cancer Research
-
Neuroscience
Cancer-induced nerve injury promotes resistance to anti-PD-1 therapy.
In Nature on 1 October 2025 by Baruch, E. N., Gleber-Netto, F. O., et al.
PubMed
Perineural invasion (PNI) is a well-established factor of poor prognosis in multiple cancer types1, yet its mechanism remains unclear. Here we provide clinical and mechanistic insights into the role of PNI and cancer-induced nerve injury (CINI) in resistance to anti-PD-1 therapy. Our study demonstrates that PNI and CINI of tumour-associated nerves are associated with poor response to anti-PD-1 therapy among patients with cutaneous squamous cell carcinoma, melanoma and gastric cancer. Electron microscopy and electrical conduction analyses reveal that cancer cells degrade the nerve fibre myelin sheets. The injured neurons respond by autonomously initiating IL-6- and type I interferon-mediated inflammation to promote nerve healing and regeneration. As the tumour grows, the CINI burden increases, and its associated inflammation becomes chronic and skews the general immune tone within the tumour microenvironment into a suppressive and exhaustive state. The CINI-driven anti-PD-1 resistance can be reversed by targeting multiple steps in the CINI signalling process: denervating the tumour, conditional knockout of the transcription factor mediating the injury signal within neurons (Atf3), knockout of interferon-α receptor signalling (Ifnar1-/-) or by combining anti-PD-1 and anti-IL-6-receptor blockade. Our findings demonstrate the direct immunoregulatory roles of CINI and its therapeutic potential.
-
-
-
Cancer Research
-
Immunology and Microbiology
-
Cell Biology
m6A-modified EHD1 controls PD-L1 endosomal trafficking to modulate immune evasion and immunotherapy responses in lung adenocarcinoma.
In Cancer Commun (Lond) on 1 October 2025 by Tian, F., Huang, J., et al.
PubMed
Eps15 homology domain (EHD) proteins, including EHD1 to EHD4, play vital roles in tumor progression. In this study, we aimed to investigate which specific EHD proteins, if any, are implicated in tumor immune evasion and immunotherapy response.
-
-
-
Immunology and Microbiology
Monocyte/macrophage-derived interleukin-15 mediates the pro-inflammatory phenotype of CD226+ B cells in type 1 diabetes.
In EBioMedicine on 1 October 2025 by Li, J., Liang, X., et al.
PubMed
Type 1 diabetes (T1D) is characterised by the autoimmune-mediated destruction of pancreatic β-cells. Although traditionally viewed as a disease dominated by T cells, recent studies have emphasised the crucial role of B cells in the development of T1D. Genome-wide association studies (GWAS) have revealed that CD226 is related to susceptibility to several autoimmune diseases, including T1D. Our recent work identified a pathogenic role of CD226+ CD8+ T cells in T1D. However, the involvement of CD226+ B cells in T1D development remains unclear.
-
-
-
Immunology and Microbiology
Intestinal fungal signatures and their impact on immune checkpoint inhibitor efficacy: a multi-cohort meta-analysis.
In NPJ Biofilms Microbiomes on 26 September 2025 by Zhang, L., Zhou, D. D., et al.
PubMed
Gut microbiota influence on the effectiveness of immune checkpoint inhibitors (ICIs), but research on fungi-an essential component of the microbiome-has been limited. This multi-cohort meta-analysis of 976 fecal metagenomes across 8 cohorts, representing melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma (RCC), identified fungal species associated with ICI efficacy. In melanoma, Rhizophagus irregularis and Debaryomyces hansenii were correlated with poor responses, whereas Aspergillus avenaceus was associated with great efficacy. In NSCLC, an increased abundance of Aspergillus pseudonomiae was associated with a favorable prognosis. Stronger bacterial-fungal interactions were observed in responders. The presence of certain fungi in fungal enterotypes, like Aspergillus or Saccharomyces, was linked to better efficacy to ICIs. Mouse models revealed Debaryomyces hansenii impaired ICI efficacy by reducing CD8+ T cells. Our findings highlight specific fungal signatures that may inform strategies to enhance ICI efficacy and encourage further research on microbial impacts on treatment outcomes.
-
-
-
Cancer Research
-
Immunology and Microbiology
CD137L promotes immune surveillance in melanoma via HLTF regulation.
In Nat Commun on 26 September 2025 by Liang, L., Zhu, L., et al.
PubMed
Immune checkpoint blockers (ICBs) have demonstrated substantial efficacy across various malignancies, yet the benefits of ICBs are limited to a subset of patients. Therefore, it is essential to identify novel therapeutic targets. By integrating multi-omics data from cohorts of patients with melanoma treated with ICBs, a positive correlation is observed between tumor CD137L expression and the efficacy of PD-1 blockade. Functionally, CD137L induction in cancer cells significantly enhances anti-tumor immunity by promoting CD8+ T cell survival, both in vivo and in vitro. Mechanistically, helicase-like transcription factor (HLTF) is identified as a pivotal transcriptional regulator of CD137L, controlling its expression through phosphorylation of serine at position 398. Therapeutically, the AMPK agonist AICAR (acadesine) as an inducer of CD137L, exhibiting synergistic effects with PD-1 or CTLA-4 blockade. In summary, our findings elucidate a mechanism controlling CD137L expression and highlight a promising combination therapy to enhance the efficacy of ICBs in melanoma. One Sentence Summary: Inducing co-stimulatory immune checkpoint CD137L expression in melanoma cells enhances T cell-mediated anti-tumor immunity.
-