InVivoPlus mouse IgG2a isotype control, unknown specificity
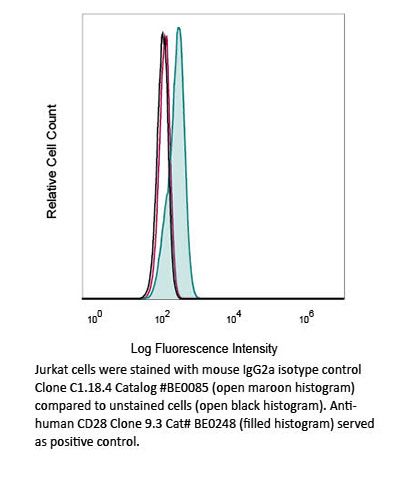
Product Description
Specifications
| Isotype | Mouse IgG2a, κ |
|---|---|
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107771 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Bulliard, Y., et al (2013). "Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies" J Exp Med 210(9): 1685-1693.
PubMed
Fc gamma receptor (FcgammaR) coengagement can facilitate antibody-mediated receptor activation in target cells. In particular, agonistic antibodies that target tumor necrosis factor receptor (TNFR) family members have shown dependence on expression of the inhibitory FcgammaR, FcgammaRIIB. It remains unclear if engagement of FcgammaRIIB also extends to the activities of antibodies targeting immunoregulatory TNFRs expressed by T cells. We have explored the requirement for activating and inhibitory FcgammaRs for the antitumor effects of antibodies targeting the TNFR glucocorticoid-induced TNFR-related protein (GITR; TNFRSF18; CD357) expressed on activated and regulatory T cells (T reg cells). We found that although FcgammaRIIB was dispensable for the in vivo efficacy of anti-GITR antibodies, in contrast, activating FcgammaRs were essential. Surprisingly, the dependence on activating FcgammaRs extended to an antibody targeting the non-TNFR receptor CTLA-4 (CD152) that acts as a negative regulator of T cell immunity. We define a common mechanism that correlated with tumor efficacy, whereby antibodies that coengaged activating FcgammaRs expressed by tumor-associated leukocytes facilitated the selective elimination of intratumoral T cell populations, particularly T reg cells. These findings may have broad implications for antibody engineering efforts aimed at enhancing the therapeutic activity of immunomodulatory antibodies.
-
Carmi, Y., et al (2015). "Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity" Nature 521(7550): 99-104.
PubMed
Whereas cancers grow within host tissues and evade host immunity through immune-editing and immunosuppression, tumours are rarely transmissible between individuals. Much like transplanted allogeneic organs, allogeneic tumours are reliably rejected by host T cells, even when the tumour and host share the same major histocompatibility complex alleles, the most potent determinants of transplant rejection. How such tumour-eradicating immunity is initiated remains unknown, although elucidating this process could provide the basis for inducing similar responses against naturally arising tumours. Here we find that allogeneic tumour rejection is initiated in mice by naturally occurring tumour-binding IgG antibodies, which enable dendritic cells (DCs) to internalize tumour antigens and subsequently activate tumour-reactive T cells. We exploited this mechanism to treat autologous and autochthonous tumours successfully. Either systemic administration of DCs loaded with allogeneic-IgG-coated tumour cells or intratumoral injection of allogeneic IgG in combination with DC stimuli induced potent T-cell-mediated antitumour immune responses, resulting in tumour eradication in mouse models of melanoma, pancreas, lung and breast cancer. Moreover, this strategy led to eradication of distant tumours and metastases, as well as the injected primary tumours. To assess the clinical relevance of these findings, we studied antibodies and cells from patients with lung cancer. T cells from these patients responded vigorously to autologous tumour antigens after culture with allogeneic-IgG-loaded DCs, recapitulating our findings in mice. These results reveal that tumour-binding allogeneic IgG can induce powerful antitumour immunity that can be exploited for cancer immunotherapy.
-
Lamere, M. W., et al (2011). "Regulation of antinucleoprotein IgG by systemic vaccination and its effect on influenza virus clearance" J Virol 85(10): 5027-5035.
PubMed
Seasonal influenza epidemics recur due to antigenic drift of envelope glycoprotein antigens and immune evasion of circulating viruses. Additionally, antigenic shift can lead to influenza pandemics. Thus, a universal vaccine that protects against multiple influenza virus strains could alleviate the continuing impact of this virus on human health. In mice, accelerated clearance of a new viral strain (cross-protection) can be elicited by prior infection (heterosubtypic immunity) or by immunization with the highly conserved internal nucleoprotein (NP). Both heterosubtypic immunity and NP-immune protection require antibody production. Here, we show that systemic immunization with NP readily accelerated clearance of a 2009 pandemic H1N1 influenza virus isolate in an antibody-dependent manner. However, human immunization with trivalent inactivated influenza virus vaccine (TIV) only rarely and modestly boosted existing levels of anti-NP IgG. Similar results were observed in mice, although the reaction could be enhanced with adjuvants, by adjusting the stoichiometry among NP and other vaccine components, and by increasing the interval between TIV prime and boost. Importantly, mouse heterosubtypic immunity that had waned over several months could be enhanced by injecting purified anti-NP IgG or by boosting with NP protein, correlating with a long-lived increase in anti-NP antibody titers. Thus, current immunization strategies poorly induce NP-immune antibody that is nonetheless capable of contributing to long-lived cross-protection. The high conservation of NP antigen and the known longevity of antibody responses suggest that the antiviral activity of anti-NP IgG may provide a critically needed component of a universal influenza vaccine.
-
Nakatsukasa, H., et al (2015). "The DNA-binding inhibitor Id3 regulates IL-9 production in CD4(+) T cells" Nat Immunol 16(10): 1077-1084.
PubMed
The molecular mechanisms by which signaling via transforming growth factor-beta (TGF-beta) and interleukin 4 (IL-4) control the differentiation of CD4(+) IL-9-producing helper T cells (TH9 cells) remain incompletely understood. We found here that the DNA-binding inhibitor Id3 regulated TH9 differentiation, as deletion of Id3 increased IL-9 production from CD4(+) T cells. Mechanistically, TGF-beta1 and IL-4 downregulated Id3 expression, and this process required the kinase TAK1. A reduction in Id3 expression enhanced binding of the transcription factors E2A and GATA-3 to the Il9 promoter region, which promoted Il9 transcription. Notably, Id3-mediated control of TH9 differentiation regulated anti-tumor immunity in an experimental melanoma-bearing model in vivo and also in human CD4(+) T cells in vitro. Thus, our study reveals a previously unrecognized TAK1-Id3-E2A-GATA-3 pathway that regulates TH9 differentiation.
Product Citations
-
IL1R2 Deficiency Unleashes Neutrophil-Mediated Antitumor Potential in Sarcoma.
In Cancer Immunol Res on 4 May 2026 by Mariancini, A., Supino, D., et al.
PubMed
Interleukin 1 (IL1) plays dual functions in cancer. It promotes cancer-related inflammation and progression but also influences leukocyte functional activation. IL1 receptor 2 (IL1R2) functions as an IL1 decoy receptor, inhibiting IL1 activity. In this study, we investigated the contribution of IL1R2 in tuning IL1-dependent effects in mouse models of cancer, including colorectal cancer, lung cancer, and primary and metastatic transplantable and chemically induced sarcoma. Even though the prominent role of IL1 is protumoral, IL1R2 deficiency was selectively associated with reduced sarcoma growth, whereas it was irrelevant in other preclinical models investigated. IL1R2 deficiency was associated with a massive infiltration of neutrophils in the tumor, neutrophilia, and increased extramedullary emergency granulopoiesis. Neutrophils were crucial for tumor control in IL1R2-deficient mice. Immunophenotypic and transcriptional profiling of sarcoma-infiltrating neutrophils revealed that IL1R2 deficiency was associated with higher expression of activation or maturation markers and gene expression reprogramming, with downregulation of pathways associated with protumoral functions. In patients with sarcoma, the IL1R2 deficiency gene signature correlated with better clinical outcomes. Thus, this study shows that IL1R2 tunes IL1-driven cancer-associated emergency granulopoiesis and neutrophil functional activation to an antitumor mode in sarcomas and reveals the antitumor potential of neutrophils in this tumor.
-
Gene Therapy with Enterovirus 3 C Protease: A Promising Strategy for Various Solid Tumors.
In Nat Commun on 8 May 2025 by Yang, X., Li, W., et al.
PubMed
Current cancer gene therapies rely primarily on antitumor immunity, but the exploration of alternative mRNA cargoes for direct antitumor effects is crucial to expand cancer gene therapies. Here we show that lipid nanoparticles (LNPs) carrying mRNA encoding a viral 3 C protease can efficiently suppress tumors by selectively inducing tumor cell apoptosis. In various solid tumor models, intracranial injection of LNPs carrying mRNA encoding the 3 C protease (3C-LNPs) significantly inhibits tumor growth and prolongs survival in glioblastoma models. Similarly, subcutaneous injection reduces tumor volume and inhibits angiogenesis in a breast cancer model, while intravenous injection inhibits tumor growth and angiogenesis and prolongs survival in hepatocellular carcinoma models. Mass spectrometry and cleavage site prediction assays identify heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) as the main target degraded by the 3 C protease. This study suggests that viral protease mRNA could be a promising broad-spectrum antitumor therapeutic.
-
CD94+ natural killer cells potentiate pulmonary ischaemia-reperfusion injury.
In Eur Respir J on 1 September 2024 by Tsao, T., Qiu, L., et al.
PubMed
Pulmonary ischaemia-reperfusion injury (IRI) is a major contributor to poor lung transplant outcomes. We recently demonstrated a central role of airway-centred natural killer (NK) cells in mediating IRI; however, there are no existing effective therapies for directly targeting NK cells in humans.
-
ITGB6 modulates resistance to anti-CD276 therapy in head and neck cancer by promoting PF4+ macrophage infiltration.
In Nat Commun on 16 August 2024 by Zhang, C., Li, K., et al.
PubMed
Enoblituzumab, an immunotherapeutic agent targeting CD276, shows both safety and efficacy in activating T cells and oligodendrocyte-like cells against various cancers. Preclinical studies and mouse models suggest that therapies targeting CD276 may outperform PD1/PD-L1 blockade. However, data from mouse models indicate a significant non-responsive population to anti-CD276 treatment, with the mechanisms of resistance still unclear. In this study, we evaluate the activity of anti-CD276 antibodies in a chemically-induced murine model of head and neck squamous cell carcinoma. Using models of induced and orthotopic carcinogenesis, we identify ITGB6 as a key gene mediating differential responses to anti-CD276 treatment. Through single-cell RNA sequencing and gene-knockout mouse models, we find that ITGB6 regulates the expression of the tumor-associated chemokine CX3CL1, which recruits and activates PF4+ macrophages that express high levels of CX3CR1. Inhibition of the CX3CL1-CX3CR1 axis suppresses the infiltration and secretion of CXCL16 by PF4+ macrophages, thereby reinvigorating cytotoxic CXCR6+ CD8+ T cells and enhancing sensitivity to anti-CD276 treatment. Further investigations demonstrate that inhibiting ITGB6 restores sensitivity to PD1 antibodies in mice resistant to anti-PD1 treatment. In summary, our research reveals a resistance mechanism associated with immune checkpoint inhibitor therapy and identifies potential targets to overcome resistance in cancer treatment.