
InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase
Product Description
Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107775 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
Goschl, L., et al. (2018). "A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis" J Autoimmun 86: 51-61.
PubMed
Multiple sclerosis (MS) is a human neurodegenerative disease characterized by the invasion of autoreactive T cells from the periphery into the CNS. Application of pan-histone deacetylase inhibitors (HDACi) ameliorates experimental autoimmune encephalomyelitis (EAE), an animal model for MS, suggesting that HDACi might be a potential therapeutic strategy for MS. However, the function of individual HDAC members in the pathogenesis of EAE is not known. In this study we report that mice with a T cell-specific deletion of HDAC1 (using the Cd4-Cre deleter strain; HDAC1-cKO) were completely resistant to EAE despite the ability of HDAC1cKO CD4(+) T cells to differentiate into Th17 cells. RNA sequencing revealed STAT1 as a prominent upstream regulator of differentially expressed genes in activated HDAC1-cKO CD4(+) T cells and this was accompanied by a strong increase in phosphorylated STAT1 (pSTAT1). This suggests that HDAC1 controls STAT1 activity in activated CD4(+) T cells. Increased pSTAT1 levels correlated with a reduced expression of the chemokine receptors Ccr4 and Ccr6, which are important for the migration of T cells into the CNS. Finally, EAE susceptibility was restored in WT:HDAC1-cKO mixed BM chimeric mice, indicating a cell-autonomous defect. Our data demonstrate a novel pathophysiological role for HDAC1 in EAE and provide evidence that selective inhibition of HDAC1 might be a promising strategy for the treatment of MS.
Clemente-Casares, X., et al. (2016). "Expanding antigen-specific regulatory networks to treat autoimmunity" Nature 530(7591): 434-440.
PubMed
Regulatory T cells hold promise as targets for therapeutic intervention in autoimmunity, but approaches capable of expanding antigen-specific regulatory T cells in vivo are currently not available. Here we show that systemic delivery of nanoparticles coated with autoimmune-disease-relevant peptides bound to major histocompatibility complex class II (pMHCII) molecules triggers the generation and expansion of antigen-specific regulatory CD4(+) T cell type 1 (TR1)-like cells in different mouse models, including mice humanized with lymphocytes from patients, leading to resolution of established autoimmune phenomena. Ten pMHCII-based nanomedicines show similar biological effects, regardless of genetic background, prevalence of the cognate T-cell population or MHC restriction. These nanomedicines promote the differentiation of disease-primed autoreactive T cells into TR1-like cells, which in turn suppress autoantigen-loaded antigen-presenting cells and drive the differentiation of cognate B cells into disease-suppressing regulatory B cells, without compromising systemic immunity. pMHCII-based nanomedicines thus represent a new class of drugs, potentially useful for treating a broad spectrum of autoimmune conditions in a disease-specific manner.
Ellis, G. T., et al. (2015). "TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection" EMBO Rep 16(9): 1203-1218.
PubMed
Streptococcus pneumoniae coinfection is a major cause of influenza-associated mortality; however, the mechanisms underlying pathogenesis or protection remain unclear. Using a clinically relevant mouse model, we identify immune-mediated damage early during coinfection as a new mechanism causing susceptibility. Coinfected CCR2(-/-) mice lacking monocytes and monocyte-derived cells control bacterial invasion better, show reduced epithelial damage and are overall more resistant than wild-type controls. In influenza-infected wild-type lungs, monocytes and monocyte-derived cells are the major cell populations expressing the apoptosis-inducing ligand TRAIL. Accordingly, anti-TRAIL treatment reduces bacterial load and protects against coinfection if administered during viral infection, but not following bacterial exposure. Post-influenza bacterial outgrowth induces a strong proinflammatory cytokine response and massive inflammatory cell infiltrate. Depletion of neutrophils or blockade of TNF-alpha facilitate bacterial outgrowth, leading to increased mortality, demonstrating that these factors aid bacterial control. We conclude that inflammatory monocytes recruited early, during the viral phase of coinfection, induce TRAIL-mediated lung damage, which facilitates bacterial invasion, while TNF-alpha and neutrophil responses help control subsequent bacterial outgrowth. We thus identify novel determinants of protection versus pathology in influenza-Streptococcus pneumoniae coinfection.
Meisen, W. H., et al. (2015). "The Impact of Macrophage- and Microglia-Secreted TNFalpha on Oncolytic HSV-1 Therapy in the Glioblastoma Tumor Microenvironment" Clin Cancer Res 21(14): 3274-3285.
PubMed
PURPOSE: Oncolytic herpes simplex viruses (oHSV) represent a promising therapy for glioblastoma (GBM), but their clinical success has been limited. Early innate immune responses to viral infection reduce oHSV replication, tumor destruction, and efficacy. Here, we characterized the antiviral effects of macrophages and microglia on viral therapy for GBM. EXPERIMENTAL DESIGN: Quantitative flow cytometry of mice with intracranial gliomas (+/-oHSV) was used to examine macrophage/microglia infiltration and activation. In vitro coculture assays of infected glioma cells with microglia/macrophages were used to test their impact on oHSV replication. Macrophages from TNFalpha-knockout mice and blocking antibodies were used to evaluate the biologic effects of TNFalpha on virus replication. TNFalpha blocking antibodies were used to evaluate the impact of TNFalpha on oHSV therapy in vivo. RESULTS: Flow-cytometry analysis revealed a 7.9-fold increase in macrophage infiltration after virus treatment. Tumor-infiltrating macrophages/microglia were polarized toward a M1, proinflammatory phenotype, and they expressed high levels of CD86, MHCII, and Ly6C. Macrophages/microglia produced significant amounts of TNFalpha in response to infected glioma cells in vitro and in vivo. Using TNFalpha-blocking antibodies and macrophages derived from TNFalpha-knockout mice, we discovered TNFalpha-induced apoptosis in infected tumor cells and inhibited virus replication. Finally, we demonstrated the transient blockade of TNFalpha from the tumor microenvironment with TNFalpha-blocking antibodies significantly enhanced virus replication and survival in GBM intracranial tumors. CONCLUSIONS: The results of these studies suggest that FDA approved TNFalpha inhibitors may significantly improve the efficacy of oncolytic virus therapy.
Park, H. J., et al. (2015). "PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells" J Immunol 194(12): 5801-5811.
PubMed
Regulatory T (Treg) cells act as terminators of T cell immuniy during acute phase of viral infection; however, their role and suppressive mechanism in chronic viral infection are not completely understood. In this study, we compared the phenotype and function of Treg cells during acute or chronic infection with lymphocytic choriomeningitis virus. Chronic infection, unlike acute infection, led to a large expansion of Treg cells and their upregulation of programmed death-1 (PD-1). Treg cells from chronically infected mice (chronic Treg cells) displayed greater suppressive capacity for inhibiting both CD8(+) and CD4(+) T cell proliferation and subsequent cytokine production than those from naive or acutely infected mice. A contact between Treg and CD8(+) T cells was necessary for the potent suppression of CD8(+) T cell immune response. More importantly, the suppression required cell-specific expression and interaction of PD-1 on chronic Treg cells and PD-1 ligand on CD8(+) T cells. Our study defines PD-1 upregulated on Treg cells and its interaction with PD-1 ligand on effector T cells as one cause for the potent T cell suppression and proposes the role of PD-1 on Treg cells, in addition to that on exhausted T cells, during chronic viral infection.
Sell, S., et al. (2015). "Control of murine cytomegalovirus infection by gammadelta T cells" PLoS Pathog 11(2): e1004481.
PubMed
Infections with cytomegalovirus (CMV) can cause severe disease in immunosuppressed patients and infected newborns. Innate as well as cellular and humoral adaptive immune effector functions contribute to the control of CMV in immunocompetent individuals. None of the innate or adaptive immune functions are essential for virus control, however. Expansion of gammadelta T cells has been observed during human CMV (HCMV) infection in the fetus and in transplant patients with HCMV reactivation but the protective function of gammadelta T cells under these conditions remains unclear. Here we show for murine CMV (MCMV) infections that mice that lack CD8 and CD4 alphabeta-T cells as well as B lymphocytes can control a MCMV infection that is lethal in RAG-1(-/-) mice lacking any T- and B-cells. gammadelta T cells, isolated from infected mice can kill MCMV infected target cells in vitro and, importantly, provide long-term protection in infected RAG-1(-/-) mice after adoptive transfer. gammadelta T cells in MCMV infected hosts undergo a prominent and long-lasting phenotypic change most compatible with the view that the majority of the gammadelta T cell population persists in an effector/memory state even after resolution of the acute phase of the infection. A clonotypically focused Vgamma1 and Vgamma2 repertoire was observed at later stages of the infection in the organs where MCMV persists. These findings add gammadelta T cells as yet another protective component to the anti-CMV immune response. Our data provide clear evidence that gammadelta T cells can provide an effective control mechanism of acute CMV infections, particularly when conventional adaptive immune mechanisms are insufficient or absent, like in transplant patient or in the developing immune system in utero. The findings have implications in the stem cell transplant setting, as antigen recognition by gammadelta T cells is not MHC-restricted and dual reactivity against CMV and tumors has been described.
Grinberg-Bleyer, Y., et al. (2015). "Cutting edge: NF-kappaB p65 and c-Rel control epidermal development and immune homeostasis in the skin" J Immunol 194(6): 2472-2476.
PubMed
Psoriasis is an inflammatory skin disease in which activated immune cells and the proinflammatory cytokine TNF are well-known mediators of pathogenesis. The transcription factor NF-kappaB is a key regulator of TNF production and TNF-induced proinflammatory gene expression, and both the psoriatic transcriptome and genetic susceptibility further implicate NF-kappaB in psoriasis etiopathology. However, the role of NF-kappaB in psoriasis remains controversial. We analyzed the function of canonical NF-kappaB in the epidermis using CRE-mediated deletion of p65 and c-Rel in keratinocytes. In contrast to animals lacking p65 or c-Rel alone, mice lacking both subunits developed severe dermatitis after birth. Consistent with its partial histological similarity to human psoriasis, this condition could be prevented by anti-TNF treatment. Moreover, regulatory T cells in lesional skin played an important role in disease remission. Our results demonstrate that canonical NF-kappaB in keratinocytes is essential for the maintenance of skin immune homeostasis and is protective against spontaneous dermatitis.
Perng, O. A., et al. (2014). "The degree of CD4+ T cell autoreactivity determines cellular pathways underlying inflammatory arthritis" J Immunol 192(7): 3043-3056.
PubMed
Although therapies targeting distinct cellular pathways (e.g., anticytokine versus anti-B cell therapy) have been found to be an effective strategy for at least some patients with inflammatory arthritis, the mechanisms that determine which pathways promote arthritis development are poorly understood. We have used a transgenic mouse model to examine how variations in the CD4(+) T cell response to a surrogate self-peptide can affect the cellular pathways that are required for arthritis development. CD4(+) T cells that are highly reactive with the self-peptide induce inflammatory arthritis that affects male and female mice equally. Arthritis develops by a B cell-independent mechanism, although it can be suppressed by an anti-TNF treatment, which prevented the accumulation of effector CD4(+) Th17 cells in the joints of treated mice. By contrast, arthritis develops with a significant female bias in the context of a more weakly autoreactive CD4(+) T cell response, and B cells play a prominent role in disease pathogenesis. In this setting of lower CD4(+) T cell autoreactivity, B cells promote the formation of autoreactive CD4(+) effector T cells (including Th17 cells), and IL-17 is required for arthritis development. These studies show that the degree of CD4(+) T cell reactivity for a self-peptide can play a prominent role in determining whether distinct cellular pathways can be targeted to prevent the development of inflammatory arthritis.
Beug, S. T., et al. (2014). "Smac mimetics and innate immune stimuli synergize to promote tumor death" Nat Biotechnol 32(2): 182-190.
PubMed
Smac mimetic compounds (SMC), a class of drugs that sensitize cells to apoptosis by counteracting the activity of inhibitor of apoptosis (IAP) proteins, have proven safe in phase 1 clinical trials in cancer patients. However, because SMCs act by enabling transduction of pro-apoptotic signals, SMC monotherapy may be efficacious only in the subset of patients whose tumors produce large quantities of death-inducing proteins such as inflammatory cytokines. Therefore, we reasoned that SMCs would synergize with agents that stimulate a potent yet safe “cytokine storm.” Here we show that oncolytic viruses and adjuvants such as poly(I:C) and CpG induce bystander death of cancer cells treated with SMCs that is mediated by interferon beta (IFN-beta), tumor necrosis factor alpha (TNF-alpha) and/or TNF-related apoptosis-inducing ligand (TRAIL). This combinatorial treatment resulted in tumor regression and extended survival in two mouse models of cancer. As these and other adjuvants have been proven safe in clinical trials, it may be worthwhile to explore their clinical efficacy in combination with SMCs.
DeBerge, M. P., et al. (2014). "Soluble, but not transmembrane, TNF-alpha is required during influenza infection to limit the magnitude of immune responses and the extent of immunopathology" J Immunol 192(12): 5839-5851.
PubMed
TNF-alpha is a pleotropic cytokine that has both proinflammatory and anti-inflammatory functions during influenza infection. TNF-alpha is first expressed as a transmembrane protein that is proteolytically processed to release a soluble form. Transmembrane TNF-alpha (memTNF-alpha) and soluble TNF-alpha (solTNF-alpha) have been shown to exert distinct tissue-protective or tissue-pathologic effects in several disease models. However, the relative contributions of memTNF-alpha or solTNF-alpha in regulating pulmonary immunopathology following influenza infection are unclear. Therefore, we performed intranasal influenza infection in mice exclusively expressing noncleavable memTNF-alpha or lacking TNF-alpha entirely and examined the outcomes. We found that solTNF-alpha, but not memTNF-alpha, was required to limit the size of the immune response and the extent of injury. In the absence of solTNF-alpha, there was a significant increase in the CD8(+) T cell response, including virus-specific CD8(+) T cells, which was due in part to an increased resistance to activation-induced cell death. We found that solTNF-alpha mediates these immunoregulatory effects primarily through TNFR1, because mice deficient in TNFR1, but not TNFR2, exhibited dysregulated immune responses and exacerbated injury similar to that observed in mice lacking solTNF-alpha. We also found that solTNF-alpha expression was required early during infection to regulate the magnitude of the CD8(+) T cell response, indicating that early inflammatory events are critical for the regulation of the effector phase. Taken together, these findings suggest that processing of memTNF-alpha to release solTNF-alpha is a critical event regulating the immune response during influenza infection.
Walsh, K. B., et al. (2014). "Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy" J Virol 88(11): 6281-6293.
PubMed
The cytokine storm is an intensified, dysregulated, tissue-injurious inflammatory response driven by cytokine and immune cell components. The cytokine storm during influenza virus infection, whereby the amplified innate immune response is primarily responsible for pulmonary damage, has been well characterized. Now we describe a novel event where virus-specific T cells induce a cytokine storm. The paramyxovirus pneumonia virus of mice (PVM) is a model of human respiratory syncytial virus (hRSV). Unexpectedly, when C57BL/6 mice were infected with PVM, the innate inflammatory response was undetectable until day 5 postinfection, at which time CD8(+) T cells infiltrated into the lung, initiating a cytokine storm by their production of gamma interferon (IFN-gamma) and tumor necrosis factor alpha (TNF-alpha). Administration of an immunomodulatory sphingosine-1-phosphate (S1P) receptor 1 (S1P1R) agonist significantly inhibited PVM-elicited cytokine storm by blunting the PVM-specific CD8(+) T cell response, resulting in diminished pulmonary disease and enhanced survival. IMPORTANCE: A dysregulated overly exuberant immune response, termed a “cytokine storm,” accompanies virus-induced acute respiratory diseases (VARV), is primarily responsible for the accompanying high morbidity and mortality, and can be controlled therapeutically in influenza virus infection of mice and ferrets by administration of sphingosine-1-phosphate 1 receptor (S1P1R) agonists. Here, two novel findings are recorded. First, in contrast to influenza infection, where the cytokine storm is initiated early by the innate immune system, for pneumonia virus of mice (PVM), a model of RSV, the cytokine storm is initiated late in infection by the adaptive immune response: specifically, by virus-specific CD8 T cells via their release of IFN-gamma and TNF-alpha. Blockading these cytokines with neutralizing antibodies blunts the cytokine storm and protects the host. Second, PVM infection is controlled by administration of an S1P1R agonist.
Weinlich, R., et al. (2013). "Protective roles for caspase-8 and cFLIP in adult homeostasis" Cell Rep 5(2): 340-348.
PubMed
Caspase-8 or cellular FLICE-like inhibitor protein (cFLIP) deficiency leads to embryonic lethality in mice due to defects in endothelial tissues. Caspase-8(-/-) and receptor-interacting protein kinase-3 (RIPK3)(-/-), but not cFLIP(-/-) and RIPK3(-/-), double-knockout animals develop normally, indicating that caspase-8 antagonizes the lethal effects of RIPK3 during development. Here, we show that the acute deletion of caspase-8 in the gut of adult mice induces enterocyte death, disruption of tissue homeostasis, and inflammation, resulting in sepsis and mortality. Likewise, acute deletion of caspase-8 in a focal region of the skin induces local keratinocyte death, tissue disruption, and inflammation. Strikingly, RIPK3 ablation rescues both phenotypes. However, acute loss of cFLIP in the skin produces a similar phenotype that is not rescued by RIPK3 ablation. TNF neutralization protects from either acute loss of caspase-8 or cFLIP. These results demonstrate that caspase-8-mediated suppression of RIPK3-induced death is required not only during development but also for adult homeostasis. Furthermore, RIPK3-dependent inflammation is dispensable for the skin phenotype.
Mohr, E., et al. (2010). "IFN-{gamma} produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine" Proc Natl Acad Sci U S A 107(40): 17292-17297.
PubMed
Alum-precipitated protein (alum protein) vaccines elicit long-lasting neutralizing antibody responses that prevent bacterial exotoxins and viruses from entering cells. Typically, these vaccines induce CD4 T cells to become T helper 2 (Th2) cells that induce Ig class switching to IgG1. We now report that CD8 T cells also respond to alum proteins, proliferating extensively and producing IFN-gamma, a key Th1 cytokine. These findings led us to question whether adoptive transfer of antigen-specific CD8 T cells alters the characteristic CD4 Th2 response to alum proteins and the switching pattern in responding B cells. To this end, WT mice given transgenic ovalbumin (OVA)-specific CD4 (OTII) or CD8 (OTI) T cells, or both, were immunized with alum-precipitated OVA. Cotransfer of antigen-specific CD8 T cells skewed switching patterns in responding B cells from IgG1 to IgG2a and IgG2b. Blocking with anti-IFN-gamma antibody largely inhibited this altered B-cell switching pattern. The transcription factor T-bet is required in B cells for IFN-gamma-dependent switching to IgG2a. By contrast, we show that this transcription factor is dispensable in B cells both for IFN-gamma-induced switching to IgG2b and for inhibition of switching to IgG1. Thus, T-bet dependence identifies distinct transcriptional pathways in B cells that regulate IFN-gamma-induced switching to different IgG isotypes.
Product Citations
-
-
Immunology and Microbiology
Dominant immune tolerance in the intestinal tract imposed by RelB-dependent migratory dendritic cells regulates protective type 2 immunity.
In Nat Commun on 23 October 2024 by Geiselhöringer, A. L., Kolland, D., et al.
PubMed
Dendritic cells (DCs) are crucial for initiating protective immune responses and have also been implicated in the generation and regulation of Foxp3+ regulatory T cells (Treg cells). Here, we show that in the lamina propria of the small intestine, the alternative NF-κB family member RelB is necessary for the differentiation of cryptopatch and isolated lymphoid follicle-associated DCs (CIA-DCs). Moreover, single-cell RNA sequencing reveals a RelB-dependent signature in migratory DCs in mesenteric lymph nodes favoring DC-Treg cell interaction including elevated expression and release of the chemokine CCL22 from RelB-deficient conventional DCs (cDCs). In line with the key role of CCL22 to facilitate DC-Treg cell interaction, RelB-deficient DCs have a selective advantage to interact with Treg cells in an antigen-specific manner. In addition, DC-specific RelB knockout animals show increased total Foxp3+ Treg cell numbers irrespective of inflammatory status. Consequently, DC-specific RelB knockout animals fail to mount protective Th2-dominated immune responses in the intestine after infection with Heligmosomoides polygyrus bakeri. Thus, RelB expression in cDCs acts as a rheostat to establish a tolerogenic set point that is maintained even during strong type 2 immune conditions and thereby is a key regulator of intestinal homeostasis.
-
-
-
Immunology and Microbiology
Interleukin-9 promotes mast cell progenitor proliferation and CCR2-dependent mast cell migration in allergic airway inflammation.
In Mucosal Immunol on 1 August 2023 by Pajulas, A., Fu, Y., et al.
PubMed
Allergic asthma is a chronic lung disease characterized by airway hyperresponsiveness and cellular infiltration that is exacerbated by immunoglobulin E-dependent mast cell (MC) activation. Interleukin-9 (IL-9) promotes MC expansion during allergic inflammation but precisely how IL-9 expands tissue MCs and promotes MC function is unclear. In this report, using multiple models of allergic airway inflammation, we show that both mature MCs (mMCs) and MC progenitors (MCp) express IL-9R and respond to IL-9 during allergic inflammation. IL-9 acts on MCp in the bone marrow and lungs to enhance proliferative capacity. Furthermore, IL-9 in the lung stimulates the mobilization of CCR2+ mMC from the bone marrow and recruitment to the allergic lung. Mixed bone marrow chimeras demonstrate that these are intrinsic effects in the MCp and mMC populations. IL-9-producing T cells are both necessary and sufficient to increase MC numbers in the lung in the context of allergic inflammation. Importantly, T cell IL-9-mediated MC expansion is required for the development of antigen-induced and MC-dependent airway hyperreactivity. Collectively, these data demonstrate that T cell IL-9 induces lung MC expansion and migration by direct effects on the proliferation of MCp and the migration of mMC to mediate airway hyperreactivity.
-
-
-
Cancer Research
-
Immunology and Microbiology
Addressing Tumor Heterogeneity by Sensitizing Resistant Cancer Cells to T cell-Secreted Cytokines.
In Cancer Discov on 4 May 2023 by Ito, Y., Pan, D., et al.
PubMed
Tumor heterogeneity is a major barrier to cancer therapy, including immunotherapy. Activated T cells can efficiently kill tumor cells following recognition of MHC class I (MHC-I)-bound peptides, but this selection pressure favors outgrowth of MHC-I-deficient tumor cells. We performed a genome-scale screen to discover alternative pathways for T cell-mediated killing of MHC-I-deficient tumor cells. Autophagy and TNF signaling emerged as top pathways, and inactivation of Rnf31 (TNF signaling) and Atg5 (autophagy) sensitized MHC-I-deficient tumor cells to apoptosis by T cell-derived cytokines. Mechanistic studies demonstrated that inhibition of autophagy amplified proapoptotic effects of cytokines in tumor cells. Antigens from apoptotic MHC-I-deficient tumor cells were efficiently cross-presented by dendritic cells, resulting in heightened tumor infiltration by IFNγ-and TNFα-producing T cells. Tumors with a substantial population of MHC-I-deficient cancer cells could be controlled by T cells when both pathways were targeted using genetic or pharmacologic approaches.
-
-
-
In vivo experiments
-
Mus musculus (Mouse)
-
Cancer Research
Bile salt hydrolase in non-enterotoxigenic Bacteroides potentiates colorectal cancer.
In Nat Commun on 10 February 2023 by Sun, L., Zhang, Y., et al.
PubMed
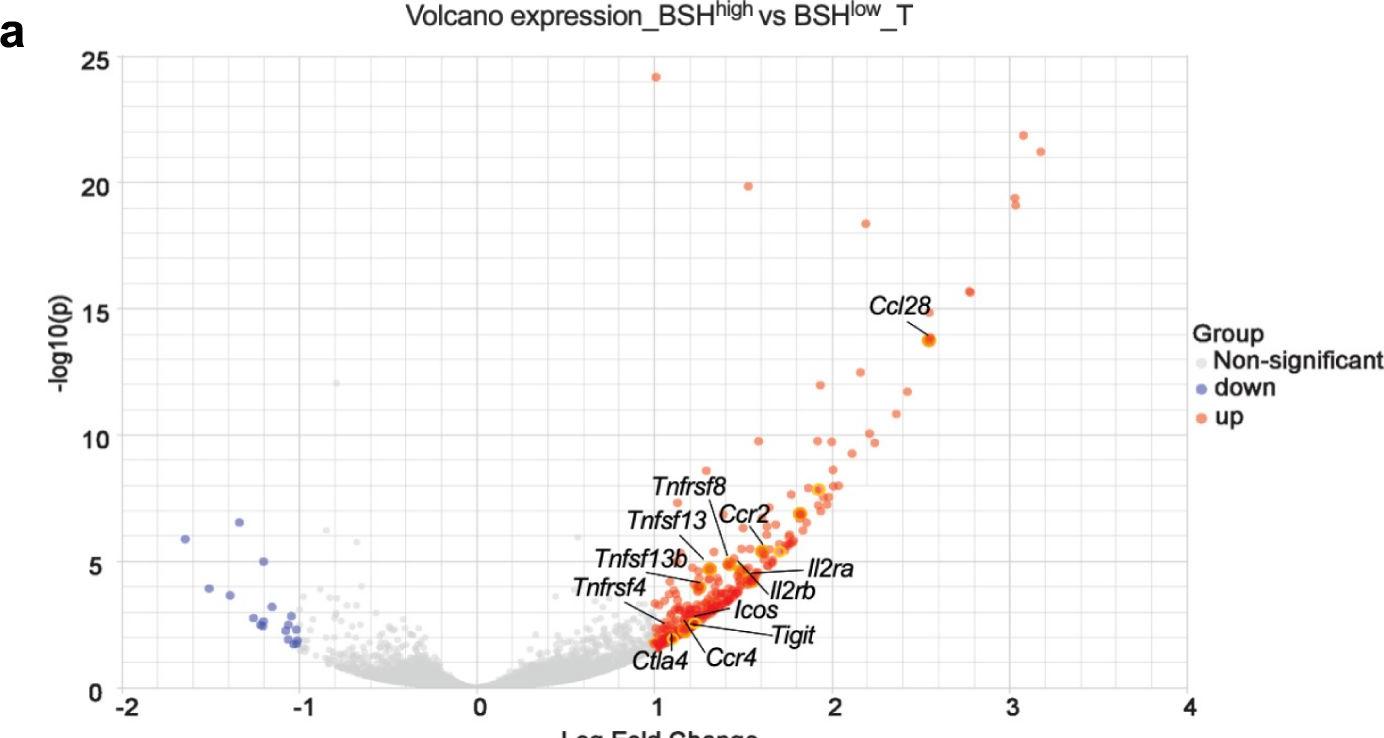
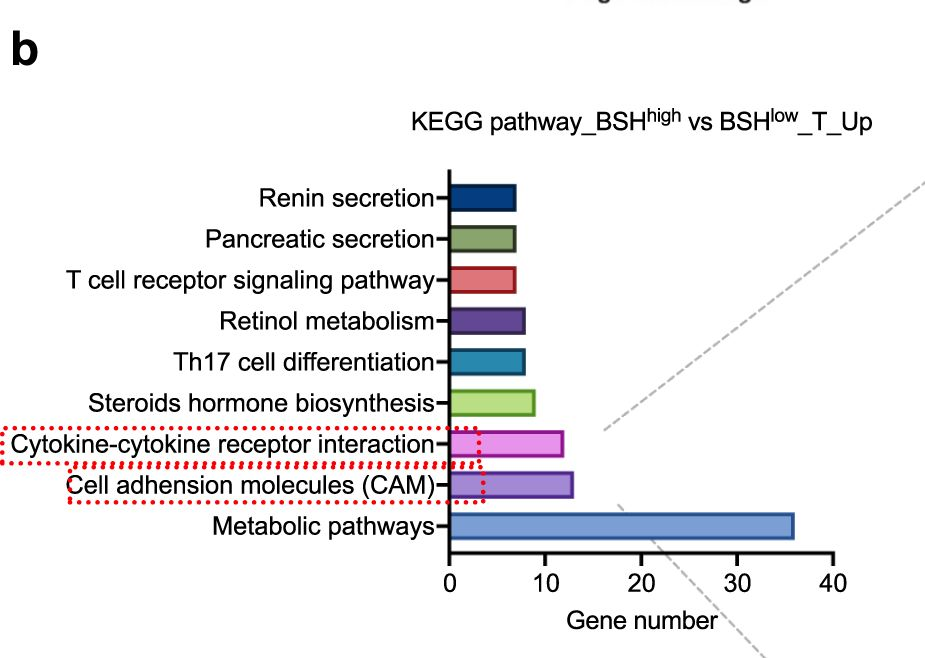
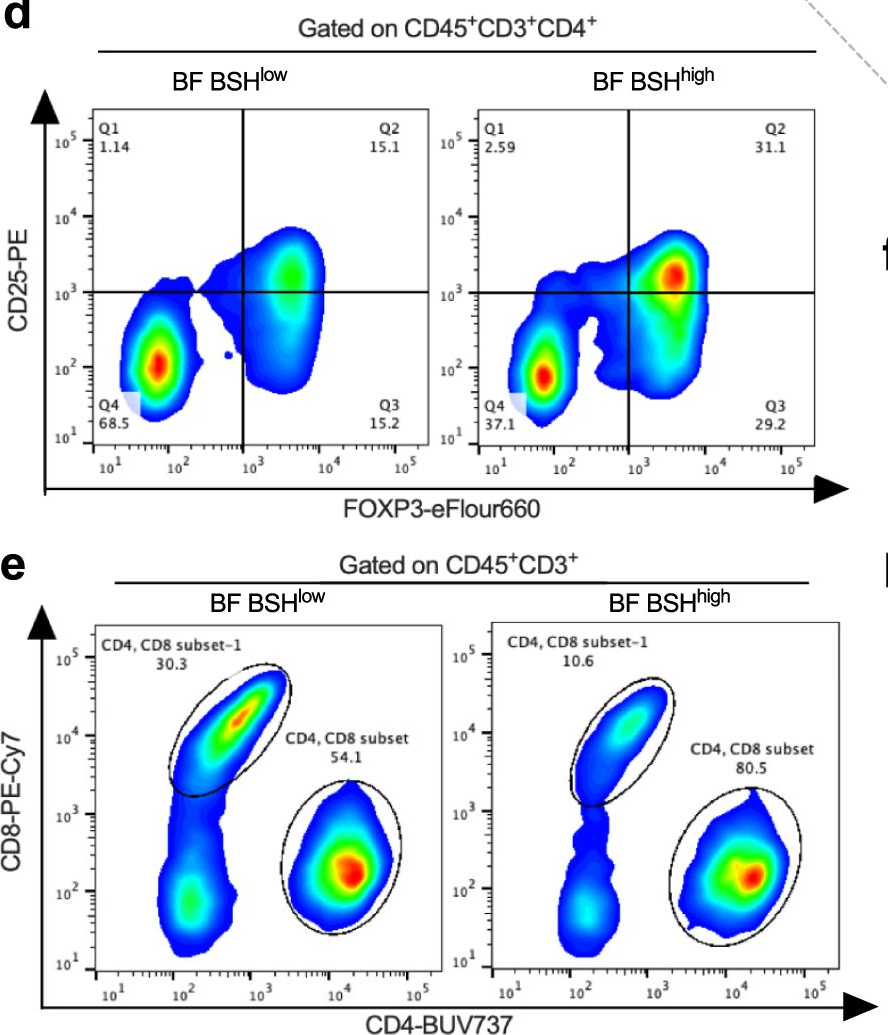
Bile salt hydrolase (BSH) in Bacteroides is considered a potential drug target for obesity-related metabolic diseases, but its involvement in colon tumorigenesis has not been explored. BSH-expressing Bacteroides is found at high abundance in the stools of colorectal cancer (CRC) patients with overweight and in the feces of a high-fat diet (HFD)-induced CRC mouse model. Colonization of B. fragilis 638R, a strain with low BSH activity, overexpressing a recombinant bsh gene from B. fragilis NCTC9343 strain, results in increased unconjugated bile acids in the colon and accelerated progression of CRC under HFD treatment. In the presence of high BSH activity, the resultant elevation of unconjugated deoxycholic acid and lithocholic acid activates the G-protein-coupled bile acid receptor, resulting in increased β-catenin-regulated chemokine (C-C motif) ligand 28 (CCL28) expression in colon tumors. Activation of the β-catenin/CCL28 axis leads to elevated intra-tumoral immunosuppressive CD25+FOXP3+ Treg cells. Blockade of the β-catenin/CCL28 axis releases the immunosuppression to enhance the intra-tumoral anti-tumor response, which decreases CRC progression under HFD treatment. Pharmacological inhibition of BSH reduces HFD-accelerated CRC progression, coincident with suppression of the β-catenin/CCL28 pathway. These findings provide insights into the pro-carcinogenetic role of Bacteroides in obesity-related CRC progression and characterize BSH as a potential target for CRC prevention and treatment.
-
-
Phase I trial of the TNF-α inhibitor certolizumab plus chemotherapy in stage IV lung adenocarcinomas.
In Nat Commun on 15 October 2022 by Paik, P., Luo, J., et al.
PubMed
We previously identified a chemotherapy-induced paracrine inflammatory loop that paradoxically mitigates the anti-tumor effect of chemotherapy and triggers metastatic propagation in breast and lung cancer models. Therefore, we sought to further validate and translate these findings into patient care by coupling the anti-TNF-α drug certolizumab pegol with standard cisplatin doublet chemotherapy. Here we first validate the anti-metastatic effect of certolizumab in a liver-metastatic Lewis Lung Carcinoma model. We then evaluate the safety, efficacy, and pharmacodynamic effects of certolizumab with cisplatin and pemetrexed in an open label Phase 1 clinical trial (NCT02120807) of eighteen adult patients with stage IV lung adenocarcinomas. The primary outcome is maximum tolerated dose. Secondary outcomes are response rate and progression-free survival (PFS); pharmacodynamic changes in blood and tumor are evaluated as a correlative outcome. There were nine partial responses among 16 patients evaluable (56%, 95% CI 30 to 80%). The median duration of response was 9.0 months (range 5.9 to 42.6 months) and median PFS was 7.1 months (95% CI 6.3 to NR). The standard 400 mg dose of certolizumab, added to cisplatin and pemetrexed, is well-tolerated and, as a correlative endpoint, demonstrates potent pharmacodynamic inhibition of peripheral cytokines associated with the paracrine inflammatory loop.
-
M-CSF supports medullary erythropoiesis and erythroid iron demand following burn injury through its activity on homeostatic iron recycling.
In Sci Rep on 24 January 2022 by Noel, J. G., Ramser, S. W., et al.
PubMed
M-CSF receptor signaling supports the development and survival of mononuclear phagocytes and is thought to play a role in post burn anemia by promoting myeloid lineage bias. We found M-CSF secretion was increased in burn patients and a murine model of post burn ACI, so we neutralized M-CSF in ACI mice to determine if erythropoiesis was improved. Instead, M-CSF blockade further impaired erythropoiesis and erythroid cells access to iron. M-CSF blockade enhanced inflammatory cytokine secretion, further increased systemic neutrophil counts, and led to tissue iron sequestration that was dependent, in part, on augmented IL-6 secretion which induced hepcidin. Deleterious effects of post burn M-CSF blockade were associated with arrest of an iron recycling gene expression signature in the liver and spleen that included Spi-C transcription factor and heme oxygenase-1, which promote heme metabolism and confer a non-inflammatory tone in macrophages. Hepatic induction of these factors in ACI mice was consistent with a recovery of ferroportin gene expression and reflected an M-CSF dependent expansion and differentiation of Spi-C+ monocytes into Kupffer cells. Together, this data indicates M-CSF secretion supports a homeostatic iron recycling program that plays a key role in the maintenance of erythroid cells access to iron following burn injury.
-
-
Biochemistry and Molecular biology
-
Cancer Research
-
Cell Biology
GCH1 induces immunosuppression through metabolic reprogramming and IDO1 upregulation in triple-negative breast cancer.
In J Immunother Cancer on 1 July 2021 by Wei, J., Wu, S. Y., et al.
PubMed
Regulatory T cells (Tregs) heavily infiltrate triple-negative breast cancer (TNBC), and their accumulation is affected by the metabolic reprogramming in cancer cells. In the present study, we sought to identify cancer cell-intrinsic metabolic modulators correlating with Tregs infiltration in TNBC.
-
-
-
Cancer Research
-
Immunology and Microbiology
A tumor-specific mechanism of Treg enrichment mediated by the integrin αvβ8.
In Sci Immunol on 26 March 2021 by Seed, R. I., Kobayashi, K., et al.
PubMed
Regulatory T cells (Tregs) that promote tumor immune evasion are enriched in certain tumors and correlate with poor prognosis. However, mechanisms for Treg enrichment remain incompletely understood. We described a mechanism for Treg enrichment in mouse and human tumors mediated by the αvβ8 integrin. Tumor cell αvβ8 bound to latent transforming growth factor-β (L-TGF-β) presented on the surface of T cells, resulting in TGF-β activation and immunosuppressive Treg differentiation in vitro. In vivo, tumor cell αvβ8 expression correlated with Treg enrichment, immunosuppressive Treg gene expression, and increased tumor growth, which was reduced in mice by αvβ8 inhibition or Treg depletion. Structural modeling and cell-based studies suggested a highly geometrically constrained complex forming between αvβ8-expressing tumor cells and L-TGF-β-expressing T cells, facilitating TGF-β activation, independent of release and diffusion, and providing limited access to TGF-β inhibitors. These findings suggest a highly localized tumor-specific mechanism for Treg enrichment.
-
-
Low-dose interleukin-2 reverses behavioral sensitization in multiple mouse models of headache disorders.
In Pain on 1 June 2020 by Zhang, J., Czerpaniak, K., et al.
PubMed
Headache disorders are highly prevalent and debilitating, with limited treatment options. Previous studies indicate that many proinflammatory immune cells contribute to headache pathophysiology. Given the well-recognized role of regulatory T (Treg) cells in maintaining immune homeostasis, we hypothesized that enhancing Treg function may be effective to treat multiple headache disorders. In a mouse model of chronic migraine, we observed that repeated nitroglycerin (NTG, a reliable trigger of migraine in patients) administration doubled the number of CD3 T cells in the trigeminal ganglia without altering the number of Treg cells, suggesting a deficiency in Treg-mediated immune homeostasis. We treated mice with low-dose interleukin-2 (ld-IL2) to preferentially expand and activate endogenous Treg cells. This not only prevented the development of NTG-induced persistent sensitization but also completely reversed the established facial skin hypersensitivity resulting from repeated NTG administration. The effect of ld-IL2 was independent of mouse sex and/or strain. Importantly, ld-IL2 treatment did not alter basal nociceptive responses, and repeated usage did not induce tolerance. The therapeutic effect of ld-IL2 was abolished by Treg depletion and was recapitulated by Treg adoptive transfer. Furthermore, treating mice with ld-IL2 1 to 7 days after mild traumatic brain injury effectively prevented as well as reversed the development of behaviors related to acute and chronic post-traumatic headache. In a model of medication overuse headache, Ld-IL2 completely reversed the cutaneous hypersensitivity induced by repeated administration of sumatriptan. Collectively, this study identifies ld-IL2 as a promising prophylactic for multiple headache disorders with a mechanism distinct from the existing treatment options.
-
-
Mus musculus (Mouse)
-
Immunology and Microbiology
Interleukin-1/-33 Signaling Pathways as Therapeutic Targets for Endometriosis.
In Front Immunol on 12 September 2019 by Kato, T., Yasuda, K., et al.
PubMed
Endometriosis is an estrogen-dependent disease with symptoms of dysmenorrhea, chronic pain, and infertility that affects 6-10% of women of reproductive age. Medical or surgical therapy, such as administration of an anti-gonadotropin or ovarian cystectomy, provide effective pain relief. However, neither therapy can be used for patients wishing to become pregnant. Despite the high morbidity, the pathogenesis of endometriosis has not been well-elucidated. Several inflammatory cytokines are reported to participate in the onset of endometriosis. Here, we examined the role of interleukin (IL)-1/IL-33 signaling in the development of endometriosis using a mouse model of endometriosis. Endometriotic lesion volume was significantly reduced in Il33-/- and Il1r1-/- mice, and almost completely suppressed in Myd88-/- mice. Mice intraperitoneally administered with an antibody against IL-1 receptor 1 (IL-1R1) or IL-33 developed limited endometriotic lesions. Oral administration of an inhibitor against IL-1R-associated kinase 4 (IRAK4), a downstream signal molecule of MyD88, also suppressed lesion formation. Furthermore, even after the development of cystic lesions the IRAK4 inhibitor prevented the enlargement of lesions. These treatments all significantly reduced cellular proliferation, shown by decreased Ki-67 expression. These results reveal that IL-1/IL-1R1, IL-33/IL-33R and associated downstream signaling molecules are involved in the pathogenesis of endometriosis, and may provide novel therapeutic targets for endometriosis.
-
-
-
Mus musculus (Mouse)
-
Immunology and Microbiology
Mucosal infection rewires TNFɑ signaling dynamics to skew susceptibility to recurrence.
In Elife on 20 August 2019 by Yu, L., O'Brien, V. P., et al.
PubMed
A mucosal infectious disease episode can render the host either more or less susceptible to recurrent infection, but the specific mechanisms that tip the balance remain unclear. We investigated this question in a mouse model of recurrent urinary tract infection and found that a prior bladder infection resulted in an earlier onset of tumor necrosis factor-alpha (TNFɑ)-mediated bladder inflammation upon subsequent bacterial challenge, relative to age-matched naive mice. However, the duration of TNFɑ signaling activation differed according to whether the first infection was chronic (Sensitized) or self-limiting (Resolved). TNFɑ depletion studies revealed that transient early-phase TNFɑ signaling in Resolved mice promoted clearance of bladder-colonizing bacteria via rapid recruitment of neutrophils and subsequent exfoliation of infected bladder cells. In contrast, sustained TNFɑ signaling in Sensitized mice prolonged damaging inflammation, worsening infection. This work reveals how TNFɑ signaling dynamics can be rewired by a prior infection to shape diverse susceptibilities to future mucosal infections.
-
-
-
In vivo experiments
-
Mus musculus (Mouse)
Fetal-derived macrophages dominate in adult mammary glands.
In Nat Commun on 17 January 2019 by Jäppinen, N., Félix, I., et al.
PubMed
Macrophages serve multiple functions including immune regulation, morphogenesis, tissue homeostasis and healing reactions. The current paradigm holds that mammary gland macrophages first arise postnatally during the prepubertal period from the bone marrow-derived monocytes. Here we delineate the origins of tissue-resident mammary gland macrophages using high-dimension phenotypic analyses, cell-fate mapping experiments, gene-deficient mice lacking selective macrophage subtypes, and antibody-based depletion strategies. We show that tissue-resident macrophages are found in mammary glands already before birth, and that the yolk sac-derived and fetal liver-derived macrophages outnumber the adult-derived macrophages in the mammary gland also in the adulthood. In addition, fetal-derived mammary gland macrophages have a characteristic phenotype, display preferential periductal and perivascular localization, and are highly active in scavenging. These findings identify fetal-derived macrophages as the predominant leukocyte type in the adult mammary gland stroma, and reveal previously unknown complexity of macrophage biology in the breast.
-
-
-
In vivo experiments
-
Mus musculus (Mouse)
-
Immunology and Microbiology
Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33.
In J Allergy Clin Immunol on 1 September 2018 by Michaudel, C., Mackowiak, C., et al.
PubMed
IL-33 plays a critical role in regulation of tissue homeostasis, injury, and repair. Whether IL-33 regulates neutrophil recruitment and functions independently of airways hyperresponsiveness (AHR) in the setting of ozone-induced lung injury and inflammation is unclear.
-
-
-
Immunology and Microbiology
CD4+ Regulatory T Cells Exert Differential Functions during Early and Late Stages of the Immune Response to Respiratory Viruses.
In J Immunol on 15 August 2018 by Rogers, M. C., Lamens, K. D., et al.
PubMed
Acute respiratory virus infection (ARI) induces CD8+ T cells with diminished cytokine production and functional impairment. The role of cellular mediators of immune impairment, specifically CD4+ regulatory T cells (Tregs), is incompletely understood in ARI. Tregs are known suppressors of effector T cell function, but whether they are detrimental or beneficial in ARI remains controversial. We show in this paper that Treg depletion leads to increased CD8+ T cell function and lower virus titer in mice infected with human metapneumovirus. We further demonstrate that Tregs play a temporal role in the immune response to human metapneumovirus and influenza: Treg depletion before infection pathologically reduces virus-specific CD8+ T cell numbers and delays virus clearance, whereas depletion 2 d postinoculation enhances CD8+ T cell functionality without reducing virus-specific CD8+ T cell numbers. Mechanistically, Treg depletion during immune priming led to impaired dendritic cell and CD8+ T cell migration. Further, early Treg depletion was associated with immune skewing toward a type 2 phenotype characterized by increased type 2 innate lymphoid cells and TH2 CD4+ T cells, which was not observed when Treg depletion was delayed until after inoculation. These results indicate that the presence of Tregs at inoculation is critical for efficient priming of the CD8+ T cell response to ARI, whereas later in infection, Tregs are dispensable for virus clearance.
-