InVivoMAb anti-mouse PD-L1 (B7-H1)
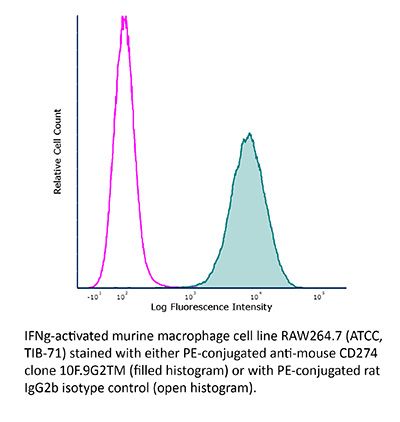
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CD274 |
| Reported Applications |
in vivo PD-L1 blockade in vitro PD-L1 blockade Immunofluorescence Immunohistochemistry (frozen) Flow cytometry Western blot in vitro Organoids/Organ-on-Chip |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949073 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Deng, L., et al (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695.
PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
-
Zander, R. A., et al (2015). "PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity" Cell Host Microbe 17(5): 628-641.
PubMed
The differentiation and protective capacity of Plasmodium-specific T cells are regulated by both positive and negative signals during malaria, but the molecular and cellular details remain poorly defined. Here we show that malaria patients and Plasmodium-infected rodents exhibit atypical expression of the co-stimulatory receptor OX40 on CD4 T cells and that therapeutic enhancement of OX40 signaling enhances helper CD4 T cell activity, humoral immunity, and parasite clearance in rodents. However, these beneficial effects of OX40 signaling are abrogated following coordinate blockade of PD-1 co-inhibitory pathways, which are also upregulated during malaria and associated with elevated parasitemia. Co-administration of biologics blocking PD-1 and promoting OX40 signaling induces excessive interferon-gamma that directly limits helper T cell-mediated support of humoral immunity and decreases parasite control. Our results show that targeting OX40 can enhance Plasmodium control and that crosstalk between co-inhibitory and co-stimulatory pathways in pathogen-specific CD4 T cells can impact pathogen clearance.
-
Hafalla, J. C., et al (2012). "The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology" PLoS Pathog 8(2): e1002504.
PubMed
The balance between pro-inflammatory and regulatory immune responses in determining optimal T cell activation is vital for the successful resolution of microbial infections. This balance is maintained in part by the negative regulators of T cell activation, CTLA-4 and PD-1/PD-L, which dampen effector responses during chronic infections. However, their role in acute infections, such as malaria, remains less clear. In this study, we determined the contribution of CTLA-4 and PD-1/PD-L to the regulation of T cell responses during Plasmodium berghei ANKA (PbA)-induced experimental cerebral malaria (ECM) in susceptible (C57BL/6) and resistant (BALB/c) mice. We found that the expression of CTLA-4 and PD-1 on T cells correlates with the extent of pro-inflammatory responses induced during PbA infection, being higher in C57BL/6 than in BALB/c mice. Thus, ECM develops despite high levels of expression of these inhibitory receptors. However, antibody-mediated blockade of either the CTLA-4 or PD-1/PD-L1, but not the PD-1/PD-L2, pathways during PbA-infection in ECM-resistant BALB/c mice resulted in higher levels of T cell activation, enhanced IFN-gamma production, increased intravascular arrest of both parasitised erythrocytes and CD8(+) T cells to the brain, and augmented incidence of ECM. Thus, in ECM-resistant BALB/c mice, CTLA-4 and PD-1/PD-L1 represent essential, independent and non-redundant pathways for maintaining T cell homeostasis during a virulent malaria infection. Moreover, neutralisation of IFN-gamma or depletion of CD8(+) T cells during PbA infection was shown to reverse the pathologic effects of regulatory pathway blockade, highlighting that the aetiology of ECM in the BALB/c mice is similar to that in C57BL/6 mice. In summary, our results underscore the differential and complex regulation that governs immune responses to malaria parasites.
-
Dietze, K. K., et al (2013). "Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads" PLoS Pathog 9(12): e1003798.
PubMed
Chronic infections with human viruses, such as HIV and HCV, or mouse viruses, such as LCMV or Friend Virus (FV), result in functional exhaustion of CD8(+) T cells. Two main mechanisms have been described that mediate this exhaustion: expression of inhibitory receptors on CD8(+) T cells and expansion of regulatory T cells (Tregs) that suppress CD8(+) T cell activity. Several studies show that blockage of one of these pathways results in reactivation of CD8(+) T cells and partial reduction in chronic viral loads. Using blocking antibodies against PD-1 ligand and Tim-3 and transgenic mice in which Tregs can be selectively ablated, we compared these two treatment strategies and combined them for the first time in a model of chronic retrovirus infection. Blocking inhibitory receptors was more efficient than transient depletion of Tregs in reactivating exhausted CD8(+) T cells and reducing viral set points. However, a combination therapy was superior to any single treatment and further augmented CD8(+) T cell responses and resulted in a sustained reduction in chronic viral loads. These results demonstrate that Tregs and inhibitory receptors are non-overlapping factors in the maintenance of chronic viral infections and that immunotherapies targeting both pathways may be a promising strategy to treat chronic infectious diseases.
Product Citations
-
A novel mRNA-based therapeutic vaccine elicits robust anti-tumor immunity against HPV-associated malignancies.
In Front Immunol on 4 May 2026 by Li, Q., Liu, Y., et al.
PubMed
Human papillomavirus (HPV) infection is strongly associated with multiple malignancies, primarily driven by the viral oncoproteins E6 and E7, which play a central role in HPV-induced malignant transformation. Although current prophylactic HPV vaccines have shown remarkable efficacy in preventing initial infections, there remains an urgent need for therapeutic vaccines targeting pre-existing HPV infections and HPV-associated malignancies.
-
Faecalibacterium prausnitzii enzyme reprograms PD-L1 trafficking and sensitizes colorectal cancer to immunotherapy in mice.
In Nat Microbiol on 1 May 2026 by Ji, S., Liu, Y., et al.
PubMed
Microbiome-host interactions can influence colorectal cancer (CRC) outcomes and the effectiveness of immunotherapy treatment, but the precise mechanisms underlying this are poorly understood. Here we analyse CRC patient cohort data and observe that Facalibacterium prausnitzii abundance in faecal samples correlates with improved CRC survival outcome and immunotherapy response. In vitro assays and experiments in azoxymethane plus dextran sulfate sodium (AOM/DSS) and Apcmin/+ mouse CRC models show that F. prausnitzii extracts have anti-tumour activity. Mass spectrometry identifies F. prausnitzii phosphoribosyl pyrophosphate synthetase (fpPRPS) as a bacterial enzyme that inhibits tumour development and promotes CD8+ T-cell responses. Mechanistically, fpPRPS depletes ATP levels in CRC cells, which then inhibits GTP-GDP exchange on Rab11a, reprogramming CRC energy metabolism. This leads to Rab11a degradation and the disruption of PD-L1 trafficking to reduce the inhibition of T-cell responses. fpPRPS inhibition of tumour progression is PD-L1-dependent. We also show that fpPRPS and anti-PD-1 treatment synergize to promote CD8+ T-cell responses and tumour control in mice. These findings suggest fpPRPS as a potential strategy for sensitizing CRC to immunotherapy.
-
Dual function of DOT1L suppresses tumor cell-intrinsic immunogenicity in hepatocellular carcinoma.
In Oncogene on 1 May 2026 by Xu, S., Gong, R., et al.
PubMed
Immune checkpoint blockade (ICB) therapy for many cancers remains limited in patients' overall response rate. Discovery and development of more effective combinatorial approaches is urgent. Here, through CRISPR/Cas9 genetic screens, we identify DOT1L as a versatile epigenetic factor that functions to suppress tumor-intrinsic immunity through a dual mechanism. Depletion of DOT1L induces the expression of transposable elements and subsequent type I interferon (IFN) response, and meanwhile lowers ZEB1 levels to further unleash the expression of immune-related genes. In turn, we demonstrate that DOT1L loss or treatment with the clinical stage inhibitor EPZ-5676 sensitizes tumors to ICB with increased immune infiltration in mice. More importantly, EPZ-5676 treatment alone is sufficient to enhance antitumor immunity in humanized mice. TCGA data analysis reveals an inverse correlation between DOT1L expression and IFN signatures across multiple cancer types. These findings provide a rationale for targeting DOT1L to improve tumor immunogenicity and overcome immunotherapy resistance.
-
Reinvigorating COTL1high NK cells via GITR signalling overcomes immune checkpoint blockade resistance in tsMHC-I-impaired tumours.
In Nat Cell Biol on 1 May 2026 by You, W., Hu, C., et al.
PubMed
Patients with impaired tumour-specific major histocompatibility complex class I (tsMHC-Iimpaired) often fail to respond to immune checkpoint blockade (ICB), presenting a major clinical challenge. However, through our multicentre investigation, we observed that a subset of patients with tsMHC-Iimpaired remains responsive to ICB, a phenomenon that has not been fully explained. Here we identify a COTL1high natural killer (NK) subset that mediates ICB responsiveness in these patients. Mechanistically, PD-L1+ macrophages coexpress GITRL and engage GITR on COTL1high NK cells, whereas PD-L1 blockade relieves the PD-1-mediated inhibition of GITR signalling and promotes NK cell activation. Activated COTL1high NK cells enhance immunological synapse stability and IFN-γ production via a metabolic-H3K27ac-RBPJ axis, thereby upregulating tsMHC-I expression and reinforcing adaptive anti-tumour immunity. Notably, GITR activation significantly enhances the sensitivity to anti-PD-L1 therapy in tsMHC-Iimpaired models. Our findings identify COTL1high NK cells as key determinants of ICB responsiveness and highlight the GITRL-GITR axis as a promising therapeutic target for tsMHC-Iimpaired tumours.