InVivoSIM anti-human PD-L1 (Atezolizumab Biosimilar)
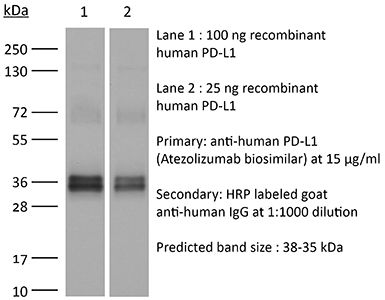
Product Description
Specifications
| Isotype | Human IgG1 |
|---|---|
| Recommended Isotype Control(s) | RecombiMAb human IgG1 (N297A) isotype control, anti-hen egg lysozyme |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Mutations | N297A |
| Immunogen | Not available or unknown |
| Reported Applications |
in vivo PD-L1 blockade Flow Cytometry Western Blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_2894730 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Sun R, Meng Z, Lee H, Offringa R, Niehrs C (2023). "ROTACs leverage signaling-incompetent R-spondin for targeted protein degradation" Cell Chem Biol 30(7):739-752.e8.
PubMed
Proteolysis-targeting chimeras (PROTACs) are an emerging technology for therapeutic intervention, but options to target cell surface proteins and receptors remain limited. Here we introduce ROTACs, bispecific WNT- and BMP-signaling-disabled R-spondin (RSPO) chimeras, which leverage the specificity of these stem cell growth factors for ZNRF3/RNF43 E3 transmembrane ligases, to target degradation of transmembrane proteins. As a proof-of-concept, we targeted the immune checkpoint protein, programmed death ligand 1 (PD-L1), a prominent cancer therapeutic target, with a bispecific RSPO2 chimera, R2PD1. The R2PD1 chimeric protein binds to PD-L1 and at picomolar concentration induces its lysosomal degradation. In three melanoma cell lines, R2PD1 induced between 50 and 90% PD-L1 protein degradation. PD-L1 degradation was strictly dependent on ZNRF3/RNF43. Moreover, R2PD1 reactivates cytotoxic T cells and inhibits tumor cell proliferation more potently than Atezolizumab. We suggest that signaling-disabled ROTACs represent a paradigm to target cell surface proteins for degradation in a range of applications.
-
Zhao Y, Caron C, Chan YY, Lee CK, Xu X, Zhang J, Masubuchi T, Wu C, Bui JD, Hui E (2023). "cis-B7:CD28 interactions at invaginated synaptic membranes provide CD28 co-stimulation and promote CD8+ T cell function and anti-tumor immunity" Immunity 56(6)
PubMed
B7 ligands (CD80 and CD86), expressed by professional antigen-presenting cells (APCs), activate the main co-stimulatory receptor CD28 on T cells in trans. However, in peripheral tissues, APCs expressing B7 ligands are relatively scarce. This raises the questions of whether and how CD28 co-stimulation occurs in peripheral tissues. Here, we report that CD8+ T cells displayed B7 ligands that interacted with CD28 in cis at membrane invaginations of the immunological synapse as a result of membrane remodeling driven by phosphoinositide-3-kinase (PI3K) and sorting-nexin-9 (SNX9). cis-B7:CD28 interactions triggered CD28 signaling through protein kinase C theta (PKCθ) and promoted CD8+ T cell survival, migration, and cytokine production. In mouse tumor models, loss of T cell-intrinsic cis-B7:CD28 interactions decreased intratumoral T cells and accelerated tumor growth. Thus, B7 ligands on CD8+ T cells can evoke cell-autonomous CD28 co-stimulation in cis in peripheral tissues, suggesting cis-signaling as a general mechanism for boosting T cell functionality.
-
Lee DH, Ahn H, Sim HI, Choi E, Choi S, Jo Y, Yun B, Song HK, Oh SJ, Denda-Nagai K, Park CS, Irimura T, Park Y, Jin HS (2023). "A CRISPR activation screen identifies MUC-21 as critical for resistance to NK and T cell-mediated cytotoxicity" J Exp Clin
PubMed
Background: Immunotherapy has significantly advanced cancer treatments, but many patients do not respond to it, partly due to immunosuppressive mechanisms used by tumor cells. These cells employ immunosuppressive ligands to evade detection and elimination by the immune system. Therefore, the discovery and characterization of novel immunosuppressive ligands that facilitate immune evasion are crucial for developing more potent anti-cancer therapies. Methods: We conducted gain-of-function screens using a CRISPRa (CRISPR activation) library that covered the entire human transmembrane sub-genome to identify surface molecules capable of hindering NK-mediated cytotoxicity. The immunosuppressive role and mechanism of MUC21 were validated using NK and T cell mediated cytotoxicity assays. Bioinformatics tools were employed to assess the clinical implications of mucin-21 (MUC21) in cancer cell immunity. Results: Our genetic screens revealed that MUC21 expression on cancer cell surfaces inhibits both the cytotoxic activity of NK cells and antibody-dependent cellular cytotoxicity, but not affecting complement-dependent cytotoxicity. Additionally, MUC21 expression hinders T cell activation by impeding antigen recognition, thereby diminishing the effectiveness of the immune checkpoint inhibitor, anti-PD-L1. Moreover, MUC21 expression suppress the antitumor function of both CAR-T cells and CAR-NK cells. Mechanistically, MUC21 facilitates immune evasion by creating steric hindrance, preventing interactions between cancer and immune cells. Bioinformatics analysis revealed elevated MUC21 expression in lung cancer, which correlated with reduced infiltration and activation of cytotoxic immune cells. Intriguingly, MUC21 expression was higher in non-small cell lung cancer (NSCLC) tumors that were non-responsive to anti-PD-(L)1 treatment compared to responsive tumors. Conclusions: These findings indicate that surface MUC21 serves as a potent immunosuppressive ligand, shielding cancer cells from NK and CD8+T cell attacks. This suggests that inhibiting MUC21 could be a promising strategy to improve cancer immunotherapy.
-
Deng R, Tian R, Li X, Xu Y, Li Y, Wang X, Li H, Wang L, Xu B, Yang D, Tang S, Xue B, Zuo C, Zhu H (2024). "ISG12a promotes immunotherapy of HBV-associated hepatocellular carcinoma through blocking TRIM21/AKT/β-catenin/PD-L1 axis" iScience 27(4):10953
PubMed
Hepatitis B virus (HBV) infection generally elicits weak type-I interferon (IFN) immune response in hepatocytes, covering the regulatory effect of IFN-stimulated genes. In this study, low level of IFN-stimulated gene 12a (ISG12a) predicted malignant transformation and poor prognosis of HBV-associated hepatocellular carcinoma (HCC), whereas high level of ISG12a indicated active NK cell phenotypes. ISG12a interacts with TRIM21 to inhibit the phosphorylation activation of protein kinase B (PKB, also known as AKT) and β-catenin, suppressing PD-L1 expression to block PD-1/PD-L1 signaling, thereby enhancing the anticancer effect of NK cells. The suppression of PD-1-deficient NK-92 cells on HBV-associated tumors was independent of ISG12a expression, whereas the anticancer effect of PD-1-expressed NK-92 cells on HBV-associated tumors was enhanced by ISG12a and treatments of atezolizumab and nivolumab. Thus, tumor intrinsic ISG12a promotes the anticancer effect of NK cells by regulating PD-1/PD-L1 signaling, presenting the significant role of innate immunity in defending against HBV-associated HCC.
Product Citations
-
PD-1 protects expanding human T cells from premature restimulation-induced cell death by modulating TCR and CD28 signaling.
In Cell Death Dis on 26 February 2026 by Lee, K. P., Elster, S., et al.
PubMed
Programmed cell death-1 (PD-1) is a co-inhibitory receptor expressed on T cells that dampens TCR and CD28 signaling in the immunological synapse. PD-1 is significantly upregulated on T cells in the tumor microenvironment, where it promotes exhaustion in the context of chronic antigen restimulation. Exhaustion renders T cells hyporesponsive and ineffectual, but potentially resistant to restimulation-induced cell death (RICD). Restimulation-induced cell death (RICD) is a critical propriocidal apoptosis program triggered in activated T cells upon robust TCR re-engagement, which serves to constrain effector T cell expansion and longevity to prevent collateral tissue damage. While the checkpoint function of PD-1 has profound implications for cancer immunotherapy, the role of PD-1 in regulating newly activated T cells remains unclear. We hypothesized that PD-1 attenuates RICD sensitivity in human effector T cells by modulating TCR signal strength. Here we show that transient upregulation of PD-1 helps to protect clonally expanding human CD4+ and CD8 + T cells from premature RICD, with only moderate protection noted in terminally-differentiated, PD-1lo effector CD8 + T cells. Restimulation of T cells with beads containing PD-L1 results in significant apoptosis resistance, dependent on PD-L1 dosage and the proximity of PD-L1 to the TCR and CD28. Interestingly, PD-L1 demonstrated a more significant RICD rescue with CD28 co-ligation as opposed to TCR engagement alone, suggesting PD-1 signaling targets both signaling pathways in this context. Furthermore, biochemical/proteomic data suggest PD-1 modulates proximal signaling downstream of both TCR and CD28 and influences the expression of specific pro/anti-apoptotic proteins that govern RICD sensitivity. Despite the original assumption of PD-1 as a programmed death-inducing protein, our research reveals that homeostatic expression of PD-1 in clonally expanding T cells confers RICD resistance that promotes T cell survival and persistence. These findings present significant implications for understanding how blocking or engaging the PD-L1:PD-1 signaling axis may influence apoptosis sensitivity in both normal and exhausted T cells to alter adaptive immune responses.
-
Probiotic-inspired hybrid nanovesicles for enhancing immune checkpoint therapy efficiency via tumor immune microenvironment modulation.
In Bioact Mater on 1 February 2026 by Wang, F., Fan, J., et al.
PubMed
Immunologically "cold" tumors, characterized by low immune cells infiltration, represent a significant obstacle to the success of immune checkpoint therapy. Intestinal microbiome therapy has emerged as a potential strategy to overcome this challenge by reprogramming the immune microenvironment. However, its clinical application is constrained by unresolved safety concerns. To address these challenges, we fused Escherichia coli-secreted outer membrane vesicle (OMV) with the macrophage membrane vector (RV) to construct hybrid nanovesicle (ROMV) and encapsulated the bacterial metabolite trimethylamine N-oxide (TMAO), forming ROMV/TMAO. ROMV/TMAO mimicked the beneficial functions of intestinal probiotics by leveraging the immunomodulatory properties of OMV and TMAO, combined with the tumor-homing capabilities of RV. In human lung cancer organoids and multiple tumor models, selective tumor targeting and accumulation of ROMV/TMAO facilitated M1 polarization of tumor-associated macrophages and enhanced CD8+ T lymphocyte infiltration, ultimately inhibiting tumor growth. When combined with ROMV/TMAO, the immune checkpoint inhibitor α-PD-L1 exhibited superior antitumor efficacy than monotherapy. This study introduces a probiotic-inspired nanotherapeutic strategy for augmenting immune checkpoint therapy outcomes while addressing microbiome therapy safety challenges.
-
Functional Role of NOXA in Hypoxia-Mediated PD-L1 Inhibitor Response in Hepatocellular Carcinoma.
In Int J Mol Sci on 16 May 2025 by Huang, M., Lan, T., et al.
PubMed
Hypoxia is a crucial characteristic of hepatocellular carcinoma (HCC) and contributes to immune resistance by upregulating PD-L1 and recruiting immunosuppressive cells. However, the molecular mechanisms of hypoxia-induced immunotherapy resistance are still unclear. The hypoxia-related immunotherapy response (IRH) genes were identified and used to develop a hypoxia risk score model to predict patient survival. The model was validated using GSE233802 and EGAD00001008128 datasets. The hypoxia risk score model including NOXA effectively stratified patients based on risk and demonstrated excellent survival predictive ability (p = 0.0236). A hypoxia-induced drug-resistant (HepG2-R) cell line was established by co-culturing HepG2 cells with Jurkat T cells under CoCl2-induced hypoxia and PD-L1 inhibitor administration. Prolonged exposure to hypoxia (48 h) in HepG2 cells significantly led to the increased hypoxia risk score (p < 0.02). The establishment of the HepG2-R cell line showed that prolonged hypoxia reduced cancer cell apoptosis, which implies potential treatment resistance. The effect of NOXA knockdown on the apoptosis of HepG2-R cells under the same co-culture conditions was examined. Under hypoxia and PD-L1 inhibitor treatment, NOXA knockdown increased the survival rate of HepG2-R cells and reduced early and late apoptosis. This indicates that NOXA plays a crucial role in apoptosis regulation and immune response in hypoxic tumors. NOXA knockdown significantly reduces apoptosis in immunotherapy-resistant cells induced by hypoxia. These findings provide important evidence that targeting NOXA may enhance immunotherapy efficacy and help overcome treatment resistance in HCC, highlighting its potential as a therapeutic target.
-
Kynurenine promotes the immune escape of colorectal cancer cells via NAT10-mediated ac4C acetylation of PD-L1.
In Clinics (Sao Paulo) on 18 April 2025 by Wang, Z., Yin, M., et al.
PubMed
This study aimed to investigate the role of kynurenine in Colorectal Cancer (CRC) and the underlying mechanism.