InVivoMAb anti-mouse PD-L1 (B7-H1)
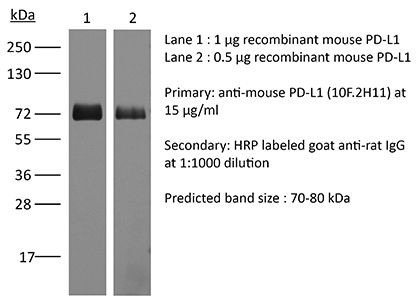
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Murine PD-L1 expressing CHO cells |
| Reported Applications |
in vivo blocking of PD-L1/CD80 (B7-1) interactions in vitro blocking of PD-L1/CD80 (B7-1) interactions ELISA Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Purification | Protein G |
| RRID | AB_2927503 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Mott, K. R., et al (2014). "Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses" PLoS One 9(1): e87617.
PubMed
CD80 plays a critical role in stimulation of T cells and subsequent control of infection. To investigate the effect of CD80 on HSV-1 infection, we constructed a recombinant HSV-1 virus that expresses two copies of the CD80 gene in place of the latency associated transcript (LAT). This mutant virus (HSV-CD80) expressed high levels of CD80 and had similar virus replication kinetics as control viruses in rabbit skin cells. In contrast to parental virus, this CD80 expressing recombinant virus replicated efficiently in immature dendritic cells (DCs). Additionally, the susceptibility of immature DCs to HSV-CD80 infection was mediated by CD80 binding to PD-L1 on DCs. This interaction also contributed to a significant increase in T cell activation. Taken together, these results suggest that inclusion of CD80 as a vaccine adjuvant may promote increased vaccine efficacy by enhancing the immune response directly and also indirectly by targeting to DC.
-
Paterson, A. M., et al (2011). "The programmed death-1 ligand 1:B7-1 pathway restrains diabetogenic effector T cells in vivo" J Immunol 187(3): 1097-1105.
PubMed
Programmed death-1 ligand 1 (PD-L1) is a coinhibitory molecule that negatively regulates multiple tolerance checkpoints. In the NOD mouse model, PD-L1 regulates the development of diabetes. PD-L1 has two binding partners, programmed death-1 and B7-1, but the significance of the PD-L1:B7-1 interaction in regulating self-reactive T cell responses is not yet clear. To investigate this issue in NOD mice, we have compared the effects of two anti-PD-L1 Abs that have different blocking activities. Anti-PD-L1 mAb 10F.2H11 sterically and functionally blocks only PD-L1:B7-1 interactions, whereas anti-PD-L1 mAb 10F.9G2 blocks both PD-L1:B7-1 and PD-L1:programmed death-1 interactions. Both Abs had potent, yet distinct effects in accelerating diabetes in NOD mice: the single-blocker 10F.2H11 mAb was more effective at precipitating diabetes in older (13-wk-old) than in younger (6- to 7-wk-old) mice, whereas the dual-blocker 10F.9G2 mAb rapidly induced diabetes in NOD mice of both ages. Similarly, 10F.2H11 accelerated diabetes in recipients of T cells from diabetic, but not prediabetic mice, whereas 10F.9G2 was effective in both settings. Both anti-PD-L1 mAbs precipitated diabetes in adoptive transfer models of CD4(+) and CD8(+) T cell-driven diabetes. Taken together, these data demonstrate that the PD-L1:B7-1 pathway inhibits potentially pathogenic self-reactive effector CD4(+) and CD8(+) T cell responses in vivo, and suggest that the immunoinhibitory functions of this pathway may be particularly important during the later phases of diabetogenesis.
-
Yang, J., et al (2011). "The novel costimulatory programmed death ligand 1/B7.1 pathway is functional in inhibiting alloimmune responses in vivo" J Immunol 187(3): 1113-1119.
PubMed
The programmed death ligand 1 (PDL1)/programmed death 1 (PD1) costimulatory pathway plays an important role in the inhibition of alloimmune responses as well as in the induction and maintenance of peripheral tolerance. It has been demonstrated recently that PDL1 also can bind B7.1 to inhibit T cell responses in vitro. Using the bm12 into B6 heart transplant model, we investigated the functional significance of this interaction in alloimmune responses in vivo. PD1 blockade unlike PDL1 blockade failed to accelerate bm12 allograft rejection, suggesting a role for an additional binding partner for PDL1 other than PD1 in transplant rejection. PDL1 blockade was able to accelerate allograft rejection in B7.2-deficient recipients but not B7.1-deficient recipients, indicating that PDL1 interaction with B7.1 was important in inhibiting rejection. Administration of the novel 2H11 anti-PDL1 mAb, which only blocks the PDL1-B7.1 interaction, aggravated chronic injury of bm12 allografts in B6 recipients. Aggravated chronic injury was associated with an increased frequency of alloreactive IFN-gamma-, IL-4-, and IL-6-producing splenocytes and a decreased percentage of regulatory T cells in the recipients. Using an in vitro cell culture assay, blockade of the interaction of PDL1 on dendritic cells with B7.1 on T cells increased IFN-gamma production from alloreactive CD4(+) T cells, whereas blockade of dendritic cell B7.1 interaction with T cell PDL1 did not. These data indicate that PDL1 interaction with B7.1 plays an important role in the inhibition of alloimmune responses in vivo and suggests a dominant direction for PDL1 and B7.1 interaction.
Product Citations
-
NPLOC4 Inhibition Remodels Tumor Microenvironment via M2-to-M1 Macrophage Reprogramming and Boosts Anti-PD-1 Response in Liver Cancer.
In Int J Biol Sci on 9 March 2026 by Gao, X., Huang, H., et al.
PubMed
The PD-1/PD-L1 axis represents a well-established immunotherapeutic target. Nevertheless, anti-PD-1/PD-L1 therapeutics have shown limited efficacy in the management of solid tumors, particularly in the context of hepatocellular carcinoma (HCC). Among the various factors contributing to the resistance to anti-PD-1/PD-L1 therapy, tumor-associated macrophages (TAMs) have attracted significant interest because of the immunosuppressive properties. NPLOC4 has been explored as an antitumor drug target. However, whether NPLOC4 functions in TAMs or immunotherapy is unclear. Here, we report a new role for NPLOC4+ TAMs in inhibiting antitumor immune responses by facilitating the proteasomal degradation of RIG-I. Clinical specimens revealed that the number of NPLOC4+ TAMs are negatively correlated with the prognosis of patients with HCC. Proteomic data and in vitro/in vivo experiments demonstrated that NPLOC4 inhibits the type I interferon pathway in TAMs, promotes M2 polarization, and suppresses CD8+ T-cell infiltration, thereby creating an immunosuppressive microenvironment in HCC. NPLOC4 can bind to RIG-I and mediate its ubiquitination-mediated degradation, thus suppressing the type I interferon pathway. Animal studies have indicated that disulfiram/copper (DSF/Cu) can target the NPLOC4 protein, and that the combination of DSF/Cu with PD-1 therapy significantly inhibits HCC growth. In conclusion, targeting NPLOC4+ TAMs can significantly increase the resistance of HCC to anti-PD-1 therapy, which makes it a promising novel immune target for HCC treatment.
-
Quantifying treatment response to a macrophage-targeted therapy in combination with immune checkpoint inhibitors after exposure to conventional chemotherapy.
In Front Immunol on 13 May 2025 by Bess, S. N., Smart, G. K., et al.
PubMed
Conventional chemotherapeutic agents, such as 5-fluorouracil (5-FU), can exert anti-tumor effects through immunogenic cell death (ICD) induction. Researchers have found hallmarks that quantify ICD (such as the translocation of HMGB1 and calreticulin). Although chemotherapeutic agents can induce ICD, they increase the expression of immune checkpoints, limiting their effectiveness. Studies have emphasized the importance of investigating the heterogeneous responses of cells co-localized in a solid tumor (macrophages, tumor cells, etc.) to ICD induction. However, these studies were performed in vivo, which limits the collection of information on cell-cell interactions due to model complexity.
-
Targeting myeloid-derived suppressor cells promotes antiparasitic T-cell immunity and enhances the efficacy of PD-1 blockade (15 words).
In Nat Commun on 27 July 2024 by Zhang, C., Wang, H., et al.
PubMed
Immune exhaustion corresponds to a loss of effector function of T cells that associates with cancer or chronic infection. Here, our objective was to decipher the mechanisms involved in the immune suppression of myeloid-derived suppressor cells (MDSCs) and to explore the potential to target these cells for immunotherapy to enhance checkpoint blockade efficacy in a chronic parasite infection. We demonstrated that programmed cell-death-1 (PD-1) expression was significantly upregulated and associated with T-cell dysfunction in advanced alveolar echinococcosis (AE) patients and in Echinococcus multilocularis-infected mice. PD-1 blockade ex vivo failed to reverse AE patients' peripheral blood T-cell dysfunction. PD-1/PD-L1 blockade or PD-1 deficiency had no significant effects on metacestode in mouse model. This was due to the inhibitory capacities of immunosuppressive granulocytic MDSCs (G-MDSCs), especially in the liver surrounding the parasite pseudotumor. MDSCs suppressed T-cell function in vitro in an indoleamine 2, 3 dioxygenase 1 (IDO1)-dependent manner. Although depleting MDSCs alone restored T-cell effector functions and led to some limitation of disease progression in E. multilocularis-infected mice, combination with PD-1 blockade was better to induce antiparasitic efficacy. Our findings provide preclinical evidence in support of targeting MDSC or combining such an approach with checkpoint blockade in patients with advanced AE. (200 words).
-
Extracellular vesicle-packaged lncRNA from cancer-associated fibroblasts promotes immune evasion by downregulating HLA-A in pancreatic cancer.
In J Extracell Vesicles on 1 July 2024 by Yao, H., Huang, C., et al.
PubMed
Pancreatic ductal adenocarcinoma (PDAC) is characterised by immune evasion that contribute to poor prognosis. Cancer-associated fibroblasts (CAFs) play a pivotal role in orchestrating the PDAC tumour microenvironment. We investigated the role of CAF-derived extracellular vesicle (EV)-packaged long non-coding RNAs (lncRNAs) in immune evasion and explored gene therapy using engineered EVs loading small interfering RNAs (siRNAs) as a potential therapeutic strategy. Our findings highlight the significance of EV-packaged lncRNA RP11-161H23.5 from CAF in promoting PDAC immune evasion by downregulating HLA-A expression, a key component of antigen presentation. Mechanistically, RP11-161H23.5 forms a complex with CNOT4, a subunit of the mRNA deadenylase CCR4-NOT complex, enhancing the degradation of HLA-A mRNA by shortening its poly(A) tail. This immune evasion mechanism compromises the anti-tumour immune response. To combat this, we propose an innovative approach utilising engineered EVs as natural and biocompatible nanocarriers for siRNA-based gene therapy and this strategy holds promise for enhancing the effectiveness of immunotherapy in PDAC. Overall, our study sheds light on the critical role of CAF-derived EV-packaged lncRNA RP11-161H23.5/CNOT4/HLA-A axis in PDAC immune evasion and presents a novel avenue for therapeutic intervention.