InVivoPlus anti-mouse CD8α
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
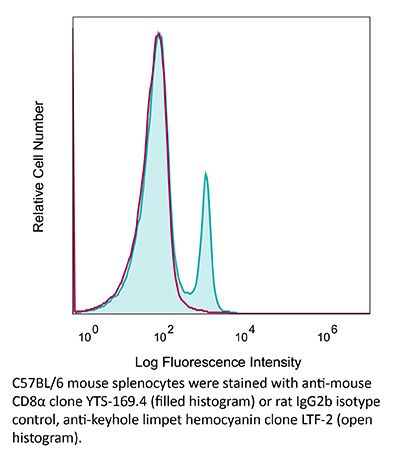
| Recommended Isotype Control(s) | InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | CBA mouse thymocytes |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950145 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Li, Z., et al (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129.
PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
-
Krupnick, A. S., et al (2014). "Central memory CD8+ T lymphocytes mediate lung allograft acceptance" J Clin Invest 124(3): 1130-1143.
PubMed
Memory T lymphocytes are commonly viewed as a major barrier for long-term survival of organ allografts and are thought to accelerate rejection responses due to their rapid infiltration into allografts, low threshold for activation, and ability to produce inflammatory mediators. Because memory T cells are usually associated with rejection, preclinical protocols have been developed to target this population in transplant recipients. Here, using a murine model, we found that costimulatory blockade-mediated lung allograft acceptance depended on the rapid infiltration of the graft by central memory CD8+ T cells (CD44(hi)CD62L(hi)CCR7+). Chemokine receptor signaling and alloantigen recognition were required for trafficking of these memory T cells to lung allografts. Intravital 2-photon imaging revealed that CCR7 expression on CD8+ T cells was critical for formation of stable synapses with antigen-presenting cells, resulting in IFN-gamma production, which induced NO and downregulated alloimmune responses. Thus, we describe a critical role for CD8+ central memory T cells in lung allograft acceptance and highlight the need for tailored approaches for tolerance induction in the lung.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
-
Vashist, N., et al (2018). "Influenza-Activated ILC1s Contribute to Antiviral Immunity Partially Influenced by Differential GITR Expression" Front Immunol 9: 505.
PubMed
Innate lymphoid cells (ILCs) represent diversified subsets of effector cells as well as immune regulators of mucosal immunity and are classified into group 1 ILCs, group 2 ILCs, and group 3 ILCs. Group 1 ILCs encompass natural killer (NK) cells and non-NK ILCs (ILC1s) and mediate their functionality via the rapid production of IFN-gamma and TNF-alpha. The current knowledge of ILC1s mainly associates them to inflammatory processes. Much less is known about their regulation during infection and their capacity to interact with cells of the adaptive immune system. The present study dissected the role of ILC1s during early influenza A virus infection, thereby revealing their impact on the antiviral response. Exploiting in vitro and in vivo H1N1 infection systems, a cross-talk of ILC1s with cells of the innate and the adaptive immunity was demonstrated, which contributes to anti-influenza immunity. A novel association of ILC1 functionality and the expression of the glucocorticoid-induced TNFR-related protein (GITR) was observed, which hints toward a so far undescribed role of GITR in regulating ILC1 responsiveness. Overexpression of GITR inhibits IFN-gamma production by ILC1s, whereas partial reduction of GITR expression can reverse this effect, thereby regulating ILC1 functionality. These new insights into ILC1 biology define potential intervention targets to modulate the functional properties of ILC1s, thus contributing toward the development of new immune interventions against influenza.
Product Citations
-
IL1R2 Deficiency Unleashes Neutrophil-Mediated Antitumor Potential in Sarcoma.
In Cancer Immunol Res on 4 May 2026 by Mariancini, A., Supino, D., et al.
PubMed
Interleukin 1 (IL1) plays dual functions in cancer. It promotes cancer-related inflammation and progression but also influences leukocyte functional activation. IL1 receptor 2 (IL1R2) functions as an IL1 decoy receptor, inhibiting IL1 activity. In this study, we investigated the contribution of IL1R2 in tuning IL1-dependent effects in mouse models of cancer, including colorectal cancer, lung cancer, and primary and metastatic transplantable and chemically induced sarcoma. Even though the prominent role of IL1 is protumoral, IL1R2 deficiency was selectively associated with reduced sarcoma growth, whereas it was irrelevant in other preclinical models investigated. IL1R2 deficiency was associated with a massive infiltration of neutrophils in the tumor, neutrophilia, and increased extramedullary emergency granulopoiesis. Neutrophils were crucial for tumor control in IL1R2-deficient mice. Immunophenotypic and transcriptional profiling of sarcoma-infiltrating neutrophils revealed that IL1R2 deficiency was associated with higher expression of activation or maturation markers and gene expression reprogramming, with downregulation of pathways associated with protumoral functions. In patients with sarcoma, the IL1R2 deficiency gene signature correlated with better clinical outcomes. Thus, this study shows that IL1R2 tunes IL1-driven cancer-associated emergency granulopoiesis and neutrophil functional activation to an antitumor mode in sarcomas and reveals the antitumor potential of neutrophils in this tumor.
-
The CHI3L1-neutrophil axis drives immune suppression and breast cancer metastatic dissemination.
In JCI Insight on 23 March 2026 by Taifour, T., Masse, A., et al.
PubMed
Immunosuppression and metastasis are critical hallmarks of breast cancer, often linked to poor patient outcomes. The secreted cytokine chitinase-3-like 1 (CHI3L1) is frequently overexpressed in breast cancer samples and promotes an immunosuppressed tumor microenvironment. Notably, CHI3L1 expression is elevated in metastatic patient samples when compared with the matched primary breast tumor. To investigate its role in breast cancer metastasis, we generated an inducible genetically engineered mouse model that overexpresses CHI3L1 in the mammary epithelium. Ectopic expression of CHI3L1 in the polyomavirus middle T (PyMT) mouse model of breast cancer suppressed antitumor immune responses, accelerated mammary tumor onset, and enhanced lung metastasis. Mechanistically, elevated CHI3L1 expression in the mammary epithelium enhanced neutrophil recruitment, which subsequently degraded the extracellular matrix and increased the number of circulating tumor cells. These findings reveal a key mechanism driving metastatic dissemination and argue that therapeutically targeting Chi3l1 could enhance antitumor immunity and suppress metastasis.
-
Targeting macrophages prevents alloantibody-mediated platelet clearance in a murine model of transfusion refractoriness.
In Blood Adv on 27 January 2026 by Rojas Jiménez, G., Angénieux, C., et al.
PubMed
HLA class I-immunized patients can experience a serious complication known as platelet transfusion refractoriness (PTR). This issue becomes especially relevant in onco-hematology departments where platelet transfusions are at the heart of patient care. Although transfusion failure is evidenced by a rapid elimination of allogeneic platelets from the recipient's bloodstream, the mechanisms behind it remain poorly characterized. The aim of this study was to better define these mechanisms to improve therapy for PTR. Using a murine model of major histocompatibility complex class I incompatibility to mimic PTR, we first established that antibodies, but not natural killer or CD8 cells, mediated platelet clearance. However, blocking Fcγ receptors with intravenous immunoglobulin or a monoclonal antibody or complement depletion did not correct refractoriness in alloimmune mice. Therefore, we investigated other alternatives beyond antibody-dependent mechanisms. Flow cytometric and microscopic analysis showed that Kupffer cells in the liver and red pulp macrophages in the spleen phagocytose allogeneic platelets during PTR. Moreover, intravital microscopy revealed allogeneic platelets retained in close interaction with macrophages in the red pulp only in alloimmune animals. Splenectomy or Kupffer cell depletion with clodronate in alloimmune mice suggested the existence of compensatory elimination mechanisms in the liver and spleen. Therefore, the simultaneous removal of both macrophage populations was an effective strategy to abrogate PTR. Our study provides an insight into the mechanisms of platelet clearance in alloimmune pathologies and opens up new perspectives for therapeutic targets.
-
A bispecific antibody targeting PD-L1/TNFR2 increases tumor targeting and enhances antitumor efficacy in colorectal cancer.
In J Immunother Cancer on 13 November 2025 by Kang, X., Qian, P., et al.
PubMed
Immune checkpoint inhibitors (ICIs) have shown limited efficacy in colorectal cancer (CRC), largely due to immunosuppressive tumor microenvironment (TME) including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs). Additionally, the off-target effects of ICIs can reduce drug accumulation in tumor tissues and lead to immune-related adverse events, further compromising their clinical utility.