InVivoMAb anti-mouse CD8α
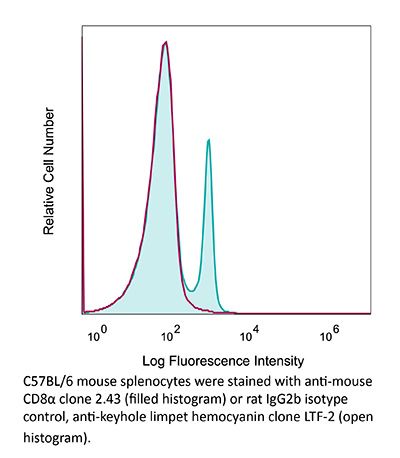
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CTL clone L3 |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1125541 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Deng, L., et al (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695.
PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Moynihan, K. D., et al (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200.
PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
-
DeBerge, M. P., et al (2014). "Soluble, but not transmembrane, TNF-alpha is required during influenza infection to limit the magnitude of immune responses and the extent of immunopathology" J Immunol 192(12): 5839-5851.
PubMed
TNF-alpha is a pleotropic cytokine that has both proinflammatory and anti-inflammatory functions during influenza infection. TNF-alpha is first expressed as a transmembrane protein that is proteolytically processed to release a soluble form. Transmembrane TNF-alpha (memTNF-alpha) and soluble TNF-alpha (solTNF-alpha) have been shown to exert distinct tissue-protective or tissue-pathologic effects in several disease models. However, the relative contributions of memTNF-alpha or solTNF-alpha in regulating pulmonary immunopathology following influenza infection are unclear. Therefore, we performed intranasal influenza infection in mice exclusively expressing noncleavable memTNF-alpha or lacking TNF-alpha entirely and examined the outcomes. We found that solTNF-alpha, but not memTNF-alpha, was required to limit the size of the immune response and the extent of injury. In the absence of solTNF-alpha, there was a significant increase in the CD8(+) T cell response, including virus-specific CD8(+) T cells, which was due in part to an increased resistance to activation-induced cell death. We found that solTNF-alpha mediates these immunoregulatory effects primarily through TNFR1, because mice deficient in TNFR1, but not TNFR2, exhibited dysregulated immune responses and exacerbated injury similar to that observed in mice lacking solTNF-alpha. We also found that solTNF-alpha expression was required early during infection to regulate the magnitude of the CD8(+) T cell response, indicating that early inflammatory events are critical for the regulation of the effector phase. Taken together, these findings suggest that processing of memTNF-alpha to release solTNF-alpha is a critical event regulating the immune response during influenza infection.
Product Citations
-
Kupffer cell calibration of T cell responses via VSIG4-CD5 interaction promotes tumor evasion.
In Nat Immunol on 1 June 2026 by Zhou, X., Liu, W., et al.
PubMed
Liver metastases can resist T cell immunotherapies, indicating an adaptation of metastatic tumors toward reduced immunogenicity in the liver. Here we show that VSIG4, an immune checkpoint molecule predominantly expressed by Kupffer cells, has an opposing function in determining the growth of liver metastases with distinct antigenicity by modulating cognate T cell antigen receptor signaling through an interaction with CD5. VSIG4-CD5 engagement impedes activation of low-affinity CD8+ T cells while enhancing responses of high-affinity CD8+ T cells by rescuing them from activation-induced cell death. This bidirectional regulation favors the outgrowth of poorly immunogenic metastatic tumor clones and fosters an immune landscape that is unfavorable to T cells as metastatic liver cancer progresses. We also show that blockade of VSIG4-CD5 interaction using a nanoantibody to VSIG4 sensitizes liver metastases to anti-PD-L1 therapy, achieving synergistic efficacy in mice. These findings provide mechanistic insights into cancer immunoediting during liver metastasis and a possible approach for treating immunologically cold tumors.
-
Bifidobacterium animalis suppresses melanoma progression and activates anti-tumor immunity by inhibiting YAP1 expression in CD8+ T cells.
In Cancer Biol Med on 6 May 2026 by Li, C., Zhang, X., et al.
PubMed
The probiotic, Bifidobacterium animalis, (B. animalis) is known to provide health benefits in humans. This study investigated the role of B. animalis in suppressing malignant melanoma progression and modulating tumor immunity.
-
A novel mRNA-based therapeutic vaccine elicits robust anti-tumor immunity against HPV-associated malignancies.
In Front Immunol on 4 May 2026 by Li, Q., Liu, Y., et al.
PubMed
Human papillomavirus (HPV) infection is strongly associated with multiple malignancies, primarily driven by the viral oncoproteins E6 and E7, which play a central role in HPV-induced malignant transformation. Although current prophylactic HPV vaccines have shown remarkable efficacy in preventing initial infections, there remains an urgent need for therapeutic vaccines targeting pre-existing HPV infections and HPV-associated malignancies.
-
Faecalibacterium prausnitzii enzyme reprograms PD-L1 trafficking and sensitizes colorectal cancer to immunotherapy in mice.
In Nat Microbiol on 1 May 2026 by Ji, S., Liu, Y., et al.
PubMed
Microbiome-host interactions can influence colorectal cancer (CRC) outcomes and the effectiveness of immunotherapy treatment, but the precise mechanisms underlying this are poorly understood. Here we analyse CRC patient cohort data and observe that Facalibacterium prausnitzii abundance in faecal samples correlates with improved CRC survival outcome and immunotherapy response. In vitro assays and experiments in azoxymethane plus dextran sulfate sodium (AOM/DSS) and Apcmin/+ mouse CRC models show that F. prausnitzii extracts have anti-tumour activity. Mass spectrometry identifies F. prausnitzii phosphoribosyl pyrophosphate synthetase (fpPRPS) as a bacterial enzyme that inhibits tumour development and promotes CD8+ T-cell responses. Mechanistically, fpPRPS depletes ATP levels in CRC cells, which then inhibits GTP-GDP exchange on Rab11a, reprogramming CRC energy metabolism. This leads to Rab11a degradation and the disruption of PD-L1 trafficking to reduce the inhibition of T-cell responses. fpPRPS inhibition of tumour progression is PD-L1-dependent. We also show that fpPRPS and anti-PD-1 treatment synergize to promote CD8+ T-cell responses and tumour control in mice. These findings suggest fpPRPS as a potential strategy for sensitizing CRC to immunotherapy.