InVivoMAb anti-mouse CD8α
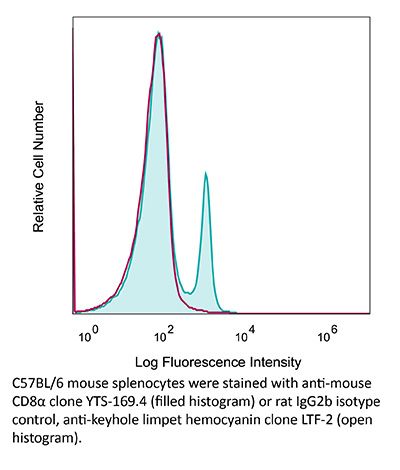
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | CBA mouse thymocytes |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950145 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Li, Z., et al (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129.
PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
-
Krupnick, A. S., et al (2014). "Central memory CD8+ T lymphocytes mediate lung allograft acceptance" J Clin Invest 124(3): 1130-1143.
PubMed
Memory T lymphocytes are commonly viewed as a major barrier for long-term survival of organ allografts and are thought to accelerate rejection responses due to their rapid infiltration into allografts, low threshold for activation, and ability to produce inflammatory mediators. Because memory T cells are usually associated with rejection, preclinical protocols have been developed to target this population in transplant recipients. Here, using a murine model, we found that costimulatory blockade-mediated lung allograft acceptance depended on the rapid infiltration of the graft by central memory CD8+ T cells (CD44(hi)CD62L(hi)CCR7+). Chemokine receptor signaling and alloantigen recognition were required for trafficking of these memory T cells to lung allografts. Intravital 2-photon imaging revealed that CCR7 expression on CD8+ T cells was critical for formation of stable synapses with antigen-presenting cells, resulting in IFN-gamma production, which induced NO and downregulated alloimmune responses. Thus, we describe a critical role for CD8+ central memory T cells in lung allograft acceptance and highlight the need for tailored approaches for tolerance induction in the lung.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
-
Vashist, N., et al (2018). "Influenza-Activated ILC1s Contribute to Antiviral Immunity Partially Influenced by Differential GITR Expression" Front Immunol 9: 505.
PubMed
Innate lymphoid cells (ILCs) represent diversified subsets of effector cells as well as immune regulators of mucosal immunity and are classified into group 1 ILCs, group 2 ILCs, and group 3 ILCs. Group 1 ILCs encompass natural killer (NK) cells and non-NK ILCs (ILC1s) and mediate their functionality via the rapid production of IFN-gamma and TNF-alpha. The current knowledge of ILC1s mainly associates them to inflammatory processes. Much less is known about their regulation during infection and their capacity to interact with cells of the adaptive immune system. The present study dissected the role of ILC1s during early influenza A virus infection, thereby revealing their impact on the antiviral response. Exploiting in vitro and in vivo H1N1 infection systems, a cross-talk of ILC1s with cells of the innate and the adaptive immunity was demonstrated, which contributes to anti-influenza immunity. A novel association of ILC1 functionality and the expression of the glucocorticoid-induced TNFR-related protein (GITR) was observed, which hints toward a so far undescribed role of GITR in regulating ILC1 responsiveness. Overexpression of GITR inhibits IFN-gamma production by ILC1s, whereas partial reduction of GITR expression can reverse this effect, thereby regulating ILC1 functionality. These new insights into ILC1 biology define potential intervention targets to modulate the functional properties of ILC1s, thus contributing toward the development of new immune interventions against influenza.
Product Citations
-
A glucocorticoid-FAS axis controls immune evasion during metastatic seeding.
In Nature on 1 May 2026 by Cassandras, M., Sánchez, X., et al.
PubMed
Metastasis is the major cause of death for patients with triple-negative breast cancer and other solid malignancies. Metastases arise from cancer cells that disseminate from the original tumour, survive systemic immune surveillance and colonize new organs1. Little is known about how initial disseminated tumour cells (DTCs) overcome anti-tumour immunity after seeding a new organ. Here we use a visible antigen in a model of triple-negative breast cancer with cognate CD8+ T cells to study the mechanisms of immune evasion in early metastatic seeding. Analysis of surviving DTCs revealed glucocorticoid receptor (GR) activation as a key driver of resistance to both CD8+ T cells and natural killer cells. Niche profiling using an optimized labelling tool identified FAS-FASL as a key pan-cytotoxic pathway against DTCs, which is repressed by GR activation. Pharmacological inhibition of GR in combination with immunotherapy reduced metastatic burden and expanded lifespan in mice. Thus, we identified a mechanism of immune evasion that operates specifically in DTCs, illustrating the unique immune-cancer interactions at this stage in the metastatic cascade. Our findings suggest that there are therapeutic opportunities to eliminate DTCs, separately from treatments aimed at primary tumours, and GR inhibition is one promising target.
-
Targeting KIF20A blocks lactylation modification to suppress immune escape in hepatocellular carcinoma.
In iScience on 17 April 2026 by Chen, S., Zhao, L., et al.
PubMed
Hepatocellular carcinoma (HCC) evades anti-PD-1 immunotherapy via an immunosuppressive microenvironment, where lactate links metabolic reprogramming to epigenetic regulation. We analyzed pan-lysine lactylation and H3K18 lactylation (H3K18la) in 89 HCC patient pairs, and validated functional mechanisms using glycolysis inhibition, HCC-CD8+ T cell co-cultures, and rescue assays. In vivo efficacy was assessed in subcutaneous and orthotopic HCC mouse models. H3K18la levels were elevated in HCC, correlating with advanced staging and poor prognosis. Lactate induced H3K18la to transcriptionally upregulate KIF20A, which stabilized the c-Myc/PD-L1 axis and suppressed cytotoxic T cell function. Combined glycolysis inhibition and anti-PD-1 therapy reversed this immunosuppression and synergistically inhibited tumor growth. This study identifies an H3K18la-KIF20A/PD-L1 axis as a key metabolic-epigenetic checkpoint, highlighting glycolysis targeting as a promising strategy to enhance anti-PD-1 responses in HCC.
-
ZBTB21 Is a Dual Suppressor of Pyroptosis and MHC-I Antigen Presentation That Promotes Tumor Immune Evasion.
In Adv Sci (Weinh) on 1 April 2026 by Zhao, L., Sheng, L., et al.
PubMed
Immune checkpoint blockade (ICB) efficacy is limited by tumor-intrinsic immune escape mechanisms. This study identifies the transcription factor ZBTB21 as a central orchestrator of dual immunosuppressive programs. ZBTB21 epigenetically silences gasdermin D (GSDMD)-dependent pyroptosis by restricting STAT1-mediated chromatin accessibility via H3K27ac modulation at the GSDMD locus. Simultaneously, it represses MHC-I antigen presentation by attenuating IRF1 expression and its transactivation capacity. Genetic ablation of ZBTB21 unleashes pyroptotic cell death and enhances tumor antigen presentation, establishing a self-reinforcing cycle that recruits and activates CD8+ T cells. This dual activation overcomes ICB resistance in murine models, while B2M deletion ablates efficacy, confirming MHC-I dependency. Pharmacological inhibition of ZBTB21 with dobutamine disrupts its DNA-binding domain, which triggers pyroptotic inflammation and MHC-I upregulation to synergize with anti-PD-1 therapy. Thus, ZBTB21 represents a druggable nexus coordinating pyroptosis resistance and antigen presentation escape, providing a combinatorial strategy to reinvigorate antitumor immunity.
-
Site-1 protease mediated GPC processing is required for persistence of LCMV Clone 13.
In Npj Viruses on 14 March 2026 by Zhou, R., Witwit, H., et al.
PubMed
Most enveloped viruses rely on furin for maturation of their surface glycoprotein. In contrast, mammarenaviruses process their glycoprotein precursor (GPC) using host site-1 protease (S1P), yet the biological implications of this unique reliance on S1P remain unclear. Here, we characterized a furin-dependent recombinant form (rCl13-RRRR) of the persistent clone 13 variant of LCMV (rCl13). Although rCl13-RRRR exhibited fitness comparable to rCl13 in cultured cells, it was highly attenuated in vivo and failed to establish persistence in immunocompetent mice. Clearance of rCl13-RRRR required interferon and CD8+ T cells, and immunization with rCl13-RRRR conferred protective immunity against a subsequent lethal LCMV challenge. Our results demonstrate that S1P-mediated processing of GPC is a key determinant of mammarenavirus fitness and immune evasion in vivo and highlight S1P as a promising and druggable target for host-directed antiviral strategies against human pathogenic mammarenaviruses.