InVivoPlus anti-mouse VEGFR-2
Product Description
Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
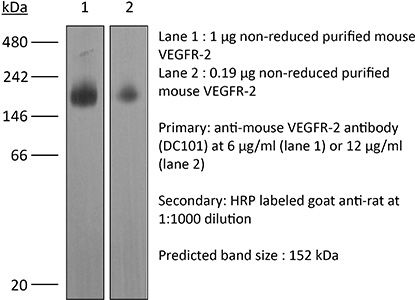
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse VEGFR-2-SEAPs soluble receptor |
| Reported Applications |
in vivo blocking of VEGF/VEGFR-2 signaling in vitro blocking of VEGFR signaling Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107766 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Arulanandam, R., et al (2015). "VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection" Cancer Cell 28(2): 210-224.
PubMed
Oncolytic viruses designed to attack malignant cells can in addition infect and destroy tumor vascular endothelial cells. We show here that this expanded tropism of oncolytic vaccinia virus to the endothelial compartment is a consequence of VEGF-mediated suppression of the intrinsic antiviral response. VEGF/VEGFR2 signaling through Erk1/2 and Stat3 leads to upregulation, nuclear localization, and activation of the transcription repressor PRD1-BF1/Blimp1. PRD1-BF1 does not contribute to the mitogenic effects of VEGF, but directly represses genes involved in type I interferon (IFN)-mediated antiviral signaling. In vivo suppression of VEGF signaling diminishes PRD1-BF1/Blimp1 expression in tumor vasculature and inhibits intravenously administered oncolytic vaccinia delivery to and consequent spread within the tumor.
-
Ding, X., et al (2015). "Distinct functions of epidermal and myeloid-derived VEGF-A in skin tumorigenesis mediated by HPV8" Cancer Res 75(2): 330-343.
PubMed
Beta human papillomaviruses (HPV) have been suspected to be carcinogenic in nonmelanoma skin cancers (NMSC), but the basis for potential viral contributions to these cancers is poorly understood. In particular, it is unresolved how HPV-infected keratinocytes escape cell-cycle control and whether their cross-talk with immune cells is critical for tumorigenesis. In nonviral preclinical models, the angiogenic cytokine VEGF-A has been identified as a critical regulator of NMSC. In this study, we dissected the contribution of epidermal versus myeloid cell-derived VEGF-A in HPV-mediated skin cancer by interbreeding an HPV8 transgenic mouse model with a conditional disruption of VEGF-A restricted to either epidermal or myeloid cells. Although only epidermal-derived VEGF-A was essential for initiation of skin tumor development, both spontaneously and UV-light triggered, both epidermal and myeloid cell-derived VEGF-A contributed to regeneration-induced tumorigenesis upon HPV8 overexpression, partly not only through a paracrine effect on endothelial cells, but also most probably through an additional autocrine effect on epidermal cells. Our findings offer new mechanistic insights into distinct functions of epidermal versus myeloid cell-derived VEGF-A during HPV-mediated tumorigenesis, with possible implications for preventing this disease.
-
Lee, H. J., et al (2015). "Inhibition of vascular endothelial growth factor A and hypoxia-inducible factor 1alpha maximizes the effects of radiation in sarcoma mouse models through destruction of tumor vasculature" Int J Radiat Oncol Biol Phys 91(3):
PubMed
PURPOSE: To examine the addition of genetic or pharmacologic inhibition of hypoxia-inducible factor 1alpha (HIF-1alpha) to radiation therapy (RT) and vascular endothelial growth factor A (VEGF-A) inhibition (ie trimodality therapy) for soft-tissue sarcoma. METHODS AND MATERIALS: Hypoxia-inducible factor 1alpha was inhibited using short hairpin RNA or low metronomic doses of doxorubicin, which blocks HIF-1alpha binding to DNA. Trimodality therapy was examined in a mouse xenograft model and a genetically engineered mouse model of sarcoma, as well as in vitro in tumor endothelial cells (ECs) and 4 sarcoma cell lines. RESULTS: In both mouse models, any monotherapy or bimodality therapy resulted in tumor growth beyond 250 mm(3) within the 12-day treatment period, but trimodality therapy with RT, VEGF-A inhibition, and HIF-1alpha inhibition kept tumors at <250 mm(3) for up to 30 days. Trimodality therapy on tumors reduced HIF-1alpha activity as measured by expression of nuclear HIF-1alpha by 87% to 95% compared with RT alone, and cytoplasmic carbonic anhydrase 9 by 79% to 82%. Trimodality therapy also increased EC-specific apoptosis 2- to 4-fold more than RT alone and reduced microvessel density by 75% to 82%. When tumor ECs were treated in vitro with trimodality therapy under hypoxia, there were significant decreases in proliferation and colony formation and increases in DNA damage (as measured by Comet assay and gammaH2AX expression) and apoptosis (as measured by cleaved caspase 3 expression). Trimodality therapy had much less pronounced effects when 4 sarcoma cell lines were examined in these same assays. CONCLUSIONS: Inhibition of HIF-1alpha is highly effective when combined with RT and VEGF-A inhibition in blocking sarcoma growth by maximizing DNA damage and apoptosis in tumor ECs, leading to loss of tumor vasculature.
-
Kizhatil, K., et al (2014). "Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process" PLoS Biol 12(7): e1001912.
PubMed
Schlemm’s canal (SC) plays central roles in ocular physiology. These roles depend on the molecular phenotypes of SC endothelial cells (SECs). Both the specific phenotype of SECs and development of SC remain poorly defined. To allow a modern and extensive analysis of SC and its origins, we developed a new whole-mount procedure to visualize its development in the context of surrounding tissues. We then applied genetic lineage tracing, specific-fluorescent reporter genes, immunofluorescence, high-resolution confocal microscopy, and three-dimensional (3D) rendering to study SC. Using these techniques, we show that SECs have a unique phenotype that is a blend of both blood and lymphatic endothelial cell phenotypes. By analyzing whole mounts of postnatal mouse eyes progressively to adulthood, we show that SC develops from blood vessels through a newly discovered process that we name “canalogenesis.” Functional inhibition of KDR (VEGFR2), a critical receptor in initiating angiogenesis, shows that this receptor is required during canalogenesis. Unlike angiogenesis and similar to stages of vasculogenesis, during canalogenesis tip cells divide and form branched chains prior to vessel formation. Differing from both angiogenesis and vasculogenesis, during canalogenesis SECs express Prox1, a master regulator of lymphangiogenesis and lymphatic phenotypes. Thus, SC development resembles a blend of vascular developmental programs. These advances define SC as a unique vessel with a combination of blood vascular and lymphatic phenotypes. They are important for dissecting its functions that are essential for ocular health and normal vision.
Product Citations
-
Intestinal lymphatic vasculature is functionally adapted to different drainage regions and is altered by helminth infection.
In J Exp Med on 1 September 2025 by Lane, J. I., Nieves-Ortiz, E., et al.
PubMed
We sought to determine whether the lymphatic vasculature functionally adapts to the organ in which it resides, such as along the gut. Duodenal lymphatic capillaries (lacteals) displayed the most discontinuous tight junction composition within the gut, resulting in a dependence on duodenal lacteals for rapid dietary lipid uptake. Duodenal helminths abrogated these features. Parallel RNA sequencing of lymphatic endothelial cells and mucosa along the intestine revealed that the transcriptomes overlapped in functional profiles. RNA sequencing also identified a putative VEGFR-2/3 signaling gradient that may explain differences in lacteal tight junctions along the small intestine at homeostasis. Transcriptionally, helminth infection triggered antimicrobial and angiogenic responses. While microbial depletion acted additively to helminths on lymphatic restructuring, glucocorticoids partially reversed helminth-induced lacteal changes. This suggests helminths induce lymphangiogenesis and associated lymphatic "zippering" via inflammation. Our study uncovers and explains the superior lipid absorption by duodenal lacteals and how it is compromised by helminths and provides transcriptional insights into lymphatic function along the gut.
-
Anti-VEGFR2 F(ab')2 drug conjugate promotes renal accumulation and glomerular repair in diabetic nephropathy.
In Nat Commun on 13 December 2023 by Liu, D., Song, Y., et al.
PubMed
Poor renal distribution of antibody-based drugs is the key factor contributing to low treatment efficiency for renal diseases and side effects. Here, we prepare F(ab')2 fragmented vascular endothelial growth factor receptor 2 antibody (anti-VEGFR2 (F(ab')2) to block VEGFR2 overactivation in diabetic nephropathy (DN). We find that the anti-VEGFR2 F(ab')2 has a higher accumulation in DN male mice kidneys than the intact VEGFR2 antibody, and simultaneously preserves the binding ability to VEGFR2. Furthermore, we develop an antibody fragment drug conjugate, anti-VEGFR2 F(ab')2-SS31, comprising the anti-VEGFR2 F(ab')2 fragment linked to the mitochondria-targeted antioxidant peptide SS31. We find that introduction of SS31 potentiates the efficacy of anti-VEGFR2 F(ab')2. These findings provide proof of concept for the premise that antibody fragment drug conjugate improves renal distribution and merits drug validation in renal disease therapy.
-
Combined inhibition of FGFR4 and VEGFR signaling enhances efficacy in FGF19 driven hepatocellular carcinoma.
In Am J Cancer Res on 12 July 2022 by Zhao, X., Joshi, J. J., et al.
PubMed
Hepatocellular carcinoma (HCC) is an aggressive liver malignancy that is difficult to treat with no approved biomarker based targeted therapies. FGF19-FGFR4 signaling blockade has been recently identified as a promising avenue for treatment of a subset of HCC patients. Using HCC relevant xenograft and PDX models, we show that Lenvatinib, an approved multi-kinase inhibitor, strongly enhanced the efficacy of FGFR4 inhibitor H3B-6527. This enhanced combination effect is not due to enhanced FGFR4 inhibition and it is likely due to cell non-autonomous VEGFR activity of Lenvatinib. This cell non-autonomous mode of action was further supported by strong in vivo combination efficacy with the mouse specific VEGFR2 antibody, DC101, which cannot cell-autonomously inhibit pathways in human xenografts. Mechanistic studies showed that the combination resulted in enhanced efficacy through increased anti-angiogenic and anti-tumorigenic activities. Overall, our results indicate that this combination can be a highly effective treatment option for FGF19 driven HCC patients, and provide preclinical validation of a combination that can be readily tested in the clinical setting.
-
Apolipoprotein E Promotes Immune Suppression in Pancreatic Cancer through NF-κB-Mediated Production of CXCL1.
In Cancer Res on 15 August 2021 by Kemp, S. B., Carpenter, E. S., et al.
PubMed
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy with few effective therapeutic options. PDAC is characterized by an extensive fibroinflammatory stroma that includes abundant infiltrating immune cells. Tumor-associated macrophages (TAM) are prevalent within the stroma and are key drivers of immunosuppression. TAMs in human and murine PDAC are characterized by elevated expression of apolipoprotein E (ApoE), an apolipoprotein that mediates cholesterol metabolism and has known roles in cardiovascular and Alzheimer's disease but no known role in PDAC. We report here that ApoE is also elevated in peripheral blood monocytes in PDAC patients, and plasma ApoE protein levels stratify patient survival. Orthotopic implantation of mouse PDAC cells into syngeneic wild-type or in ApoE-/- mice showed reduced tumor growth in ApoE-/- mice. Histologic and mass cytometric (CyTOF) analysis of these tumors showed an increase in CD8+ T cells in tumors in ApoE-/- mice. Mechanistically, ApoE induced pancreatic tumor cell expression of Cxcl1 and Cxcl5, known immunosuppressive factors, through LDL receptor and NF-κB signaling. Taken together, this study reveals a novel immunosuppressive role of ApoE in the PDAC microenvironment. SIGNIFICANCE: This study shows that elevated apolipoprotein E in PDAC mediates immune suppression and high serum apolipoprotein E levels correlate with poor patient survival.See related commentary by Sherman, p. 4186.