InVivoMAb anti-mouse CTLA-4 (CD152)
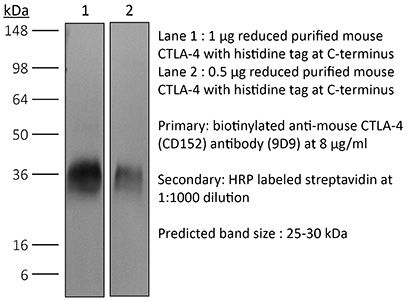
Product Description
Specifications
| Isotype | Mouse IgG2b |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG2b isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vivo CTLA-4 neutralization Western blot in vivo intra-tumoral regulatory T cell depletionin vitro Organoids/Organ-on-Chip |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_10949609 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Redmond, W. L., et al (2014). "Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity" Cancer Immunol Res 2(2): 142-153.
PubMed
Ligation of the TNF receptor family costimulatory molecule OX40 (CD134) with an agonist anti-OX40 monoclonal antibody (mAb) enhances antitumor immunity by augmenting T-cell differentiation as well as turning off the suppressive activity of the FoxP3(+)CD4(+) regulatory T cells (Treg). In addition, antibody-mediated blockade of the checkpoint inhibitor CTLA-4 releases the “brakes” on T cells to augment tumor immunotherapy. However, monotherapy with these agents has limited therapeutic benefit particularly against poorly immunogenic murine tumors. Therefore, we examined whether the administration of agonist anti-OX40 therapy in the presence of CTLA-4 blockade would enhance tumor immunotherapy. Combined anti-OX40/anti-CTLA-4 immunotherapy significantly enhanced tumor regression and the survival of tumor-bearing hosts in a CD4 and CD8 T cell-dependent manner. Mechanistic studies revealed that the combination immunotherapy directed the expansion of effector T-bet(high)/Eomes(high) granzyme B(+) CD8 T cells. Dual immunotherapy also induced distinct populations of Th1 [interleukin (IL)-2, IFN-gamma], and, surprisingly, Th2 (IL-4, IL-5, and IL-13) CD4 T cells exhibiting increased T-bet and Gata-3 expression. Furthermore, IL-4 blockade inhibited the Th2 response, while maintaining the Th1 CD4 and effector CD8 T cells that enhanced tumor-free survival. These data demonstrate that refining the global T-cell response during combination immunotherapy can further enhance the therapeutic efficacy of these agents.
-
Dai, M., et al (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138.
PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
-
Wei, H., et al (2013). "Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin" PLoS One 8(12): e84927.
PubMed
90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>There is an urgent need for improved therapy for advanced ovarian carcinoma, which may be met by administering immune-modulatory monoclonal antibodies (mAbs) to generate a tumor-destructive immune response. Using the ID8 mouse ovarian cancer model, we investigated the therapeutic efficacy of various mAb combinations in mice with intraperitoneal (i.p.) tumor established by transplanting 3 x 10(6) ID8 cells 10 days previously. While most of the tested mAbs were ineffective when given individually or together, the data confirm our previous finding that 2 i.p. injections of a combination of anti-CD137 with anti-PD-1 mAbs doubles overall survival. Mice treated with this mAb combination have a significantly increased frequency and total number of CD8(+) T cells both in the peritoneal lavage and spleens, and these cells are functional as demonstrated by antigen-specific cytolytic activity and IFN-gamma production. While administration of anti-CD137 mAb as a single agent similarly increases CD8(+) T cells, these have no functional activity, which may be attributed to up-regulation of co-inhibitory PD-1 and TIM-3 molecules induced by CD137. Addition of the anti-cancer drug cisplatin to the 2 mAb combination increased overall survival >90 days (and was probably curative) by a mechanism which included a systemic CD8(+) T cell response with tumor specificity and immunological memory. Strikingly, combined treatment of cisplatin and CD137/PD-1 mAb also gave rise to the long-term survival of mice with established TC1 lung tumors. A similar combination of the 2 mAbs and cisplatin should be considered for clinical ‘translation’.
Product Citations
-
Rescue of Tolerant CD8+ T Cells during Cancer Immunotherapy with IL2:Antibody Complexes.
In Cancer Immunology Research on 1 December 2016 by Klevorn, L. E., Berrien-Elliott, M. M., et al.
PubMed
Interleukin-2 (IL2) was among the earliest reagents used for cancer immunotherapy due to its ability to support the survival and function of tumor-reactive T cells. However, treatment with IL2 is accompanied by off-target toxicity and low response rates in patients. In mouse models, these issues are largely overcome when IL2 is administered as a cytokine/antibody complex (IL2c). The complex has a longer serum half-life and can be designed for preferential cytokine delivery to specific cells of interest. Early studies showed IL2c could boost antitumor immunity in mice by activating tumor-reactive CD8+ T cells. But such functional T cells are often limited in the tumor microenvironment, where instead unresponsive tolerant T cells are eventually eliminated by apoptosis, representing a major obstacle to the success of cancer immunotherapy. We found that IL2c treatment rescued tumor-specific CD8+ T cells from a state of established tolerance, providing effective immunotherapy in tumor-bearing mice. Expression of the transcription factor T-bet was necessary to drive intratumoral IFNγ production and effector activity by T cells rescued with IL2c. Furthermore, IL2c promoted T-bet expression in human CD4+ and CD8+ T cells in humanized tumor-bearing mice, but also increased the frequency of Foxp3+ regulatory T cells. Our study reveals a novel role for IL2c as a powerful immunotherapeutic reagent capable of reversing tolerance in tumor-reactive T cells, and provides the first evidence that IL2c influences human T cells in vivo, highlighting the translational potential to modulate human antitumor immune responses. Cancer Immunol Res; 4(12); 1016-26. ©2016 AACR. ©2016 American Association for Cancer Research.
-
Characterization of HER2-Positive Murine Breast Cancer Models for Investigating HER2-Targeted Therapy and Immunotherapy.
In Cancers (Basel) on 19 March 2026 by Lu, Y., Lee, B. P., et al.
PubMed
Background/Objectives: Human epidermal growth factor receptor 2 (HER2)-positive breast cancer is linked to poorer overall survival and a higher risk of brain metastases compared to HER2-negative breast cancer. Current preclinical studies lack robust HER2+ metastatic syngeneic mouse models for investigating targeted and immunomodulatory therapies. This study aims to develop effective HER2+ mouse models to investigate response dynamics to HER2-targeted therapy and immunotherapy. Methods: The human HER2 gene (WT or mutant p.A775_G776insYVMA, GFP-tagged at the C-terminus) was introduced into triple-negative breast cancer (TNBC) mouse mammary carcinoma cells with known metastatic potential (4T1 and EO771) via lentiviral transduction. HER2 expression and phosphorylation were analyzed using Western blotting and immunohistochemistry. Tumors were treated with HER2-targeted therapy (trastuzumab and tucatinib), immune checkpoint blockade (anti-PD-1 and anti-CTLA-4), and anti-HER2 antibody-drug conjugate (ADC) to evaluate treatment efficacy. Metastatic potential was assessed with brain fluorescence imaging. Statistical analysis included ANOVA and Kaplan-Meier tests. Results: Newly established lines demonstrated expression of HER2+, with HER2YVMA lines showing higher phosphorylation than HER2WT lines. Cells were tumorigenic, demonstrating in vivo tumor take rates at 100% for 4T1-HER2 and 15-30% for EO771-HER2. HER2 overexpression led to a 30% increase in spontaneous brain metastasis in the 4T1-HER2 models. Trastuzumab alone did not reduce primary tumor size but significantly reduced brain GFP signal by 17% ± 8% and 26% ± 7% in the 4T1-HER2WT and 4T1-HER2YVMA models, respectively. Combinational therapies with anti-HER2 therapy and immune checkpoint blockade effectively suppressed primary tumor growth and prolonged survival in EO771-HER2YVMA model. T-Dxd, but not T-DM1, demonstrated partial treatment response in the EO771-HER2WT model. Conclusions: HER2+ syngeneic tumor models were developed that spontaneously metastasize to the brain and demonstrate variable responses to immunotherapies and ADCs. These models are valuable for advancing molecular imaging modalities for HER2+ brain metastasis, studying blood-brain barrier penetration of HER2-targeted drugs, and exploring the combination of therapies, including immunotherapy.
-
Targeting the Mapk13-Tcf1-Slc7a5 Axis via One-Carbon Metabolic Regulation to Prevent Chronic Allograft Vasculopathy.
In Adv Sci (Weinh) on 1 March 2026 by Yi, W., Wu, D., et al.
PubMed
Chronic allograft vasculopathy (CAV) is driven in part by stem-like CD4+ T cells, but how these cells sustain their progenitor programs during chronic rejection remains unclear. Here, a metabolic-epigenetic axis is identified in which Mapk13 phosphorylates Tcf1 at T289, enabling Tcf1 to activate the amino acid transporter Slc7a5 and enhance methionine uptake. This rewires one-carbon metabolism and increases H3K4me3 enrichment at the Tcf7 locus, thereby maintaining stem-like CD4+ T cells within rejecting grafts. Disruption of this circuit-via genetic deletion of Mapk13 or Slc7a5, or through dietary methionine restriction-reduces Tcf1+ CD4+ T cell stemness and prevents CAV in mouse models. These findings reveal the Mapk13-Tcf1-Slc7a5 axis as a critical metabolic dependency of pathogenic T cells and highlight one-carbon metabolism as a promising target to promote long-term graft survival.
-
Ubiquitination-directed cytosolic DNA degradation governs cGAS-STING-mediated immune response to DNA damage.
In Cancer Cell on 9 February 2026 by Li, L., Ye, Q., et al.
PubMed
Activation of cGAS-STING signaling in cancer cells requires cytosolic DNA produced by intrinsic or treatment-induced DNA damage. However, clinical efforts to exploit this pathway to improve immunotherapy have yielded limited success, highlighting gaps in understanding the link between DNA damage and immunotherapy. Here, we identify ubiquitination-directed cytosolic DNA degradation as a critical determinant for cGAS-STING activation following DNA damage. Mechanistically, the cytosolic DNA exonuclease TREX1 is degraded by the E3 ubiquitin ligase SPOP but is reversely stabilized by the deubiquitinase USP7. Cancer-associated SPOP mutations or USP7 overexpression elevate TREX1 levels, promoting cytosolic DNA degradation and impairing cGAS-STING-mediated immune activation. Notably, elevated USP7 expression correlates with reduced tumor-infiltrating lymphocytes and accelerated disease progression in patients undergoing chemoradiotherapy. Furthermore, USP7 inhibitors reduce TREX1 levels and restore immune responses following radiation. These findings elucidate the mechanisms linking DNA damage to immune activation and highlight USP7 inhibitors as potential enhancers of radioimmunotherapy.