InVivoMAb anti-human PD-1 (CD279)
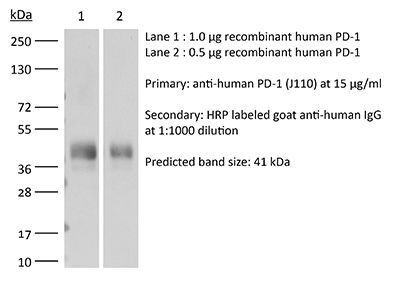
Product Description
Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
in vivo PD-1 blockade in humanized mice Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950168 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Sanlorenzo, M., et al (2018). "BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti-PD-1 Antibody" Clin Cancer Res 24(14): 3377-3385.
PubMed
Purpose: BRAF and MEK inhibitors (BRAF/MEKi) favor melanoma-infiltrating lymphocytes, providing the rationale for current combinatorial trials with anti-PD-1 antibody. A portion of melanoma cells may express PD-1, and anti-PD-1 antibody could have a direct antitumor effect. Here, we explore whether BRAF/MEKi modulate rates of PD-1(+) melanoma cells, supporting an additional-lymphocyte-independent-basis for their therapeutic combination with anti-PD-1 antibody.Experimental Design: With data mining and flow cytometry, we assessed PD-1, PD-L1/2 expression on melanoma cell lines (CCLE, N = 61; validation cell lines, N = 7) and melanoma tumors (TCGA, N = 214). We explored in vitro how BRAF/MEKi affect rates of PD-1(+), PD-L1/2(+) melanoma cells, and characterized the proliferative and putative stemness features of PD-1(+) melanoma cells. We tested the functional lymphocyte-independent effect of anti-PD-1 antibody alone and in combination with BRAF/MEKi in vitro and in an in vivo immunodeficient murine model.Results: PD-1 is consistently expressed on a small subset of melanoma cells, but PD-1(+) cells increase to relevant rates during BRAF/MEKi treatment [7.3% (5.6-14.2) vs. 1.5% (0.7-3.2), P = 0.0156; N = 7], together with PD-L2(+) melanoma cells [8.5% (0.0-63.0) vs. 1.5% (0.2-43.3), P = 0.0312; N = 7]. PD-1(+) cells proliferate less than PD-1(-) cells (avg. 65% less; t = 7 days) and are preferentially endowed with stemness features. In vivo, the direct anti-melanoma activity of PD-1 blockage as monotherapy was negligible, but its association with BRAF/MEKi significantly delayed the development of drug resistance and tumor relapse.Conclusions: BRAF/MEKi increase the rates of PD-1(+) melanoma cells that may sustain tumor relapse, providing a lymphocyte-independent rationale to explore combinatory strategies with anti-PD-1 antibody. Clin Cancer Res; 24(14); 3377-85.
-
Yamane, H., et al (2015). "Programmed cell death protein 1 and programmed death-ligand 1 are expressed on the surface of some small-cell lung cancer lines" Am J Cancer Res 5(4): 1553-1557.
PubMed
INTRODUCTION: Programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) play a major role in suppressing the immune system during the formation of the PD-1/PD-L1 pathway, which transmits an inhibitory signal to reduce T cell activity. PD-L1 is often expressed in various malignant tumors. In contrast, PD-1 is generally observed in activated lymphocytes and myeloid-derived dendritic cells. Of the malignant cells, only Jurkat cells under special conditions and angioimmunoblastic T-cell lymphoma tissue cells express PD-1 on their surface. METHODS: To clarify whether the PD-1/PD-L1 pathway participates in the immunotolerance of small-cell lung cancer (SCLC) cells, we examined the expressions of PD-1 and PD-L1 on the cell surface of SCLC cell lines using flow cytometry and reverse transcription polymerase chain reaction. RESULTS: Among the four SCLC cell lines examined, only SBC-3 expressed both PD-1 and PD-L1. CONCLUSIONS: We demonstrated that both PD-1 and PD-L1 molecules were co-expressed on the surface of SCLC cells. Although the biological implications of this remain unclear, we speculate that PD-1 and its ligand on the SCLC cells may participate in the growth inhibition of tumor cells as reported in cytotoxic T cells.
-
Bennett, F., et al (2003). "Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses" J Immunol 170(2): 711-7
PubMed
The program death 1 (PD-1) receptor and its ligands, PD-1 ligand (PD-L)1 and PD-L2, define a novel regulatory pathway with potential inhibitory effects on T, B, and monocyte responses. In the present study, we show that human CD4(+) T cells express PD-1, PD-L1, and PD-L2 upon activation, and Abs to the receptor can be agonists or antagonists of the pathway. Under optimal conditions of stimulation, ICOS but not CD28 costimulation can be prevented by PD-1 engagement. IL-2 levels induced by costimulation are critical in determining the outcome of the PD-1 engagement. Thus, low to marginal IL-2 levels produced upon ICOS costimulation account for the greater sensitivity of this pathway to PD-1-mediated inhibition. Interestingly, exogenous IL-2, IL-7, and IL-15 but not IL-4 and IL-21 can rescue PD-1 inhibition, suggesting that among these cytokines only those that activate STAT5 can rescue PD-1 inhibition. As STAT5 has been implicated in the maintenance of IL-2Ralpha expression, these results suggest that IL-7 and IL-15 restore proliferation under conditions of PD-1 engagement by enhancing high-affinity IL-2R expression and hence, IL-2 responsiveness.
Product Citations
-
BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti-PD-1 Antibody.
In Clinical Cancer Research on 15 July 2018 by Sanlorenzo, M., Vujic, I., et al.
PubMed
Purpose: BRAF and MEK inhibitors (BRAF/MEKi) favor melanoma-infiltrating lymphocytes, providing the rationale for current combinatorial trials with anti-PD-1 antibody. A portion of melanoma cells may express PD-1, and anti-PD-1 antibody could have a direct antitumor effect. Here, we explore whether BRAF/MEKi modulate rates of PD-1+ melanoma cells, supporting an additional-lymphocyte-independent-basis for their therapeutic combination with anti-PD-1 antibody.Experimental Design: With data mining and flow cytometry, we assessed PD-1, PD-L1/2 expression on melanoma cell lines (CCLE, N = 61; validation cell lines, N = 7) and melanoma tumors (TCGA, N = 214). We explored in vitro how BRAF/MEKi affect rates of PD-1+, PD-L1/2+ melanoma cells, and characterized the proliferative and putative stemness features of PD-1+ melanoma cells. We tested the functional lymphocyte-independent effect of anti-PD-1 antibody alone and in combination with BRAF/MEKi in vitro and in an in vivo immunodeficient murine model.Results: PD-1 is consistently expressed on a small subset of melanoma cells, but PD-1+ cells increase to relevant rates during BRAF/MEKi treatment [7.3% (5.6-14.2) vs. 1.5% (0.7-3.2), P = 0.0156; N = 7], together with PD-L2+ melanoma cells [8.5% (0.0-63.0) vs. 1.5% (0.2-43.3), P = 0.0312; N = 7]. PD-1+ cells proliferate less than PD-1- cells (avg. 65% less; t = 7 days) and are preferentially endowed with stemness features. In vivo, the direct anti-melanoma activity of PD-1 blockage as monotherapy was negligible, but its association with BRAF/MEKi significantly delayed the development of drug resistance and tumor relapse.Conclusions: BRAF/MEKi increase the rates of PD-1+ melanoma cells that may sustain tumor relapse, providing a lymphocyte-independent rationale to explore combinatory strategies with anti-PD-1 antibody. Clin Cancer Res; 24(14); 3377-85. ©2018 AACR. ©2018 American Association for Cancer Research.
-
Inactivating LATS2 Variation Drives Tumor Progression and Resistance to Anti-PD-1 Therapy in Intrahepatic Cholangiocarcinoma.
In Clin Mol Hepatol on 18 March 2026 by Xu, Y., Liu, K. X., et al.
PubMed
Recurrence is a major factor limiting the long-term survival of patients with intrahepatic cholangiocarcinoma (ICC). The molecular characteristics and potential therapeutic targets in ICC remain largely undefined.
-
PD-1 endocytosis unleashes the cytolytic potential of checkpoint blockade in tumor immunity.
In Cell Rep on 26 November 2024 by Ben Saad, E., Oroya, A., et al.
PubMed
PD-1 immune checkpoint blockade (ICB) is a key cancer treatment. While blocking PD-1 binding to ligand is known, the role of internalization in enhancing ICB efficacy is less explored. Our study reveals that PD-1 internalization helps unlock ICB's full potential in cancer immunotherapy. Anti-PD-1 induces 50%-60% surface PD-1 internalization from human and mouse cells, leaving low to intermediate levels of resistant receptors. Complexes then appear in early and late endosomes. Both CD4 and CD8 T cells, especially CD8+ effectors, are affected. Nivolumab outperforms pembrolizumab in human T cells, while PD-1 internalization requires crosslinking by bivalent antibody. While mono- and bivalent anti-PD-1 inhibit tumor growth with CD8 tumor-infiltrating cells expressing increased granzyme B, bivalent antibody is more effective where the combination of steric blockade and endocytosis induces greater CD8+ T cell tumor infiltration and the expression of the cytolytic pore protein, perforin. Our findings highlight an ICB mechanism that combines steric blockade and PD-1 endocytosis for optimal checkpoint immunotherapy.
-
AXL targeting restores PD-1 blockade sensitivity of STK11/LKB1 mutant NSCLC through expansion of TCF1+ CD8 T cells.
In Cell Rep Med on 15 March 2022 by Li, H., Liu, Z., et al.
PubMed
Mutations in STK11/LKB1 in non-small cell lung cancer (NSCLC) are associated with poor patient responses to immune checkpoint blockade (ICB), and introduction of a Stk11/Lkb1 (L) mutation into murine lung adenocarcinomas driven by mutant Kras and Trp53 loss (KP) resulted in an ICB refractory syngeneic KPL tumor. Mechanistically this occurred because KPL mutant NSCLCs lacked TCF1-expressing CD8 T cells, a phenotype recapitulated in human STK11/LKB1 mutant NSCLCs. Systemic inhibition of Axl results in increased type I interferon secretion from dendritic cells that expanded tumor-associated TCF1+PD-1+CD8 T cells, restoring therapeutic response to PD-1 ICB in KPL tumors. This was observed in syngeneic immunocompetent mouse models and in humanized mice bearing STK11/LKB1 mutant NSCLC human tumor xenografts. NSCLC-affected individuals with identified STK11/LKB1 mutations receiving bemcentinib and pembrolizumab demonstrated objective clinical response to combination therapy. We conclude that AXL is a critical targetable driver of immune suppression in STK11/LKB1 mutant NSCLC.