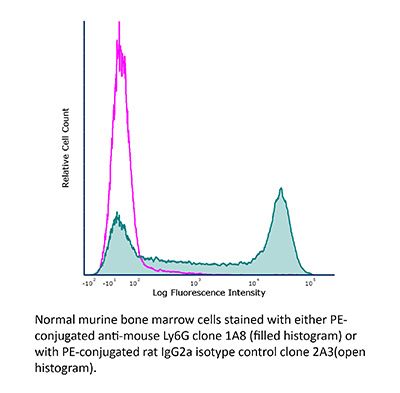
InVivoPlus rat IgG2a isotype control, anti-trinitrophenol
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by DLS |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107769 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
in vitro T cell stimulation/activation
Xiao, N., et al (2014). "The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells" Nat Immunol 15(7): 657-666.
PubMed
Follicular helper T cells (T(FH) cells) are responsible for effective B cell-mediated immunity, and Bcl-6 is a central factor for the differentiation of T(FH) cells. However, the molecular mechanisms that regulate the induction of T(FH) cells remain unclear. Here we found that the E3 ubiquitin ligase Itch was essential for the differentiation of T(FH) cells, germinal center responses and immunoglobulin G (IgG) responses to acute viral infection. Itch acted intrinsically in CD4(+) T cells at early stages of T(FH) cell development. Itch seemed to act upstream of Bcl-6 expression, as Bcl-6 expression was substantially impaired in Itch(-/-) cells, and the differentiation of Itch(-/-) T cells into T(FH) cells was restored by enforced expression of Bcl-6. Itch associated with the transcription factor Foxo1 and promoted its ubiquitination and degradation. The defective T(FH) differentiation of Itch(-/-) T cells was rectified by deletion of Foxo1. Thus, our results indicate that Itch acts as an essential positive regulator in the differentiation of T(FH) cells.
Bamboat, Z. M., et al (2010). "Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury" Hepatology 51(2): 621-632.
PubMed
Endogenous ligands such as high-mobility group box 1 (HMGB1) and nucleic acids are released by dying cells and bind Toll-like receptors (TLRs). Because TLR9 sits at the interface of microbial and sterile inflammation by detecting both bacterial and endogenous DNA, we investigated its role in a model of segmental liver ischemia-reperfusion (I/R) injury. Mice were subjected to 1 hour of ischemia and 12 hours of reperfusion before assessment of liver injury, cytokines, and reactive oxygen species (ROS). Wild-type (WT) mice treated with an inhibitory cytosine-guanosine dinucleotide (iCpG) sequence and TLR9(-/-) mice had markedly reduced serum alanine aminotransferase (ALT) and inflammatory cytokines after liver I/R. Liver damage was mediated by bone marrow-derived cells because WT mice transplanted with TLR9(-/-) bone marrow were protected from hepatic I/R injury. Injury in WT mice partly depended on TLR9 signaling in neutrophils, which enhanced production of ROS, interleukin-6 (IL-6), and tumor necrosis factor (TNF). In vitro, DNA released from necrotic hepatocytes increased liver nonparenchymal cell (NPC) and neutrophil cytokine secretion through a TLR9-dependent mechanism. Inhibition of both TLR9 and HMGB1 caused maximal inflammatory cytokine suppression in neutrophil cultures and conferred even greater protection from I/R injury in vivo. CONCLUSION: TLR9 serves as an endogenous sensor of tissue necrosis that exacerbates the innate immune response during liver I/R. Combined blockade of TLR9 and HMGB1 represents a clinically relevant, novel approach to limiting I/R injury.
in vivo NK cell depletion
Flow Cytometry
in vivo CD8+ T cell depletion
Flow Cytometry
in vivo IFNγ neutralization
Walsh, K. B., et al (2014). "Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy" J Virol 88(11): 6281-6293.
PubMed
The cytokine storm is an intensified, dysregulated, tissue-injurious inflammatory response driven by cytokine and immune cell components. The cytokine storm during influenza virus infection, whereby the amplified innate immune response is primarily responsible for pulmonary damage, has been well characterized. Now we describe a novel event where virus-specific T cells induce a cytokine storm. The paramyxovirus pneumonia virus of mice (PVM) is a model of human respiratory syncytial virus (hRSV). Unexpectedly, when C57BL/6 mice were infected with PVM, the innate inflammatory response was undetectable until day 5 postinfection, at which time CD8(+) T cells infiltrated into the lung, initiating a cytokine storm by their production of gamma interferon (IFN-gamma) and tumor necrosis factor alpha (TNF-alpha). Administration of an immunomodulatory sphingosine-1-phosphate (S1P) receptor 1 (S1P1R) agonist significantly inhibited PVM-elicited cytokine storm by blunting the PVM-specific CD8(+) T cell response, resulting in diminished pulmonary disease and enhanced survival. IMPORTANCE: A dysregulated overly exuberant immune response, termed a “cytokine storm,” accompanies virus-induced acute respiratory diseases (VARV), is primarily responsible for the accompanying high morbidity and mortality, and can be controlled therapeutically in influenza virus infection of mice and ferrets by administration of sphingosine-1-phosphate 1 receptor (S1P1R) agonists. Here, two novel findings are recorded. First, in contrast to influenza infection, where the cytokine storm is initiated early by the innate immune system, for pneumonia virus of mice (PVM), a model of RSV, the cytokine storm is initiated late in infection by the adaptive immune response: specifically, by virus-specific CD8 T cells via their release of IFN-gamma and TNF-alpha. Blockading these cytokines with neutralizing antibodies blunts the cytokine storm and protects the host. Second, PVM infection is controlled by administration of an S1P1R agonist.
in vivo activation of 4-1BB
in vivo CTLA-4 neutralization
in vivo CD19 neutralization
Flow Cytometry
Dai, M., et al (2015). "Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies" Clin Cancer Res 21(5): 1127-1138.
PubMed
PURPOSE: Immunomodulatory mAbs can treat cancer, but cures are rare except for small tumors. Our objective was to explore whether the therapeutic window increases by combining mAbs with different modes of action and injecting them into tumors. EXPERIMENTAL DESIGN: Combinations of mAbs to CD137/PD-1/CTLA-4 or CD137/PD-1/CTLA-4/CD19 were administrated intratumorally to mice with syngeneic tumors (B16 and SW1 melanoma, TC1 lung carcinoma), including tumors with a mean surface of approximately 80 mm(2). Survival and tumor growth were assessed. Immunologic responses were evaluated using flow cytometry and qRT-PCR. RESULTS: More than 50% of tumor-bearing mice had complete regression and long-term survival after tumor injection with mAbs recognizing CD137/PD-1/CTLA-4/CD19 with similar responses in three models. Intratumoral injection was more efficacious than intraperitoneal injection in causing rejection also of untreated tumors in the same mice. The three-mAb combination could also induce regression, but was less efficacious. There were few side effects, and therapy-resistant tumors were not observed. Transplanted tumor cells rapidly caused a Th2 response with increased CD19 cells. Successful therapy shifted this response to the Th1 phenotype with decreased CD19 cells and increased numbers of long-term memory CD8 effector cells and T cells making IFNgamma and TNFalpha. CONCLUSIONS: Intratumoral injection of mAbs recognizing CD137/PD-1/CTLA-4/CD19 can eradicate established tumors and reverse a Th2 response with tumor-associated CD19 cells to Th1 immunity, whereas a combination lacking anti-CD19 is less effective. There are several human cancers for which a similar approach may provide clinical benefit.
in vivo macrophage depletion
in vivo CD40 activation
in vivo LAG-3 neutralization
in vivo blocking of ICOS/ICOSL signaling
Flow Cytometry
Bauche, D., et al (2018). "LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis" Immunity 49(2): 342-352 e345.
PubMed
Interleukin-22 (IL-22)-producing group 3 innate lymphoid cells (ILC3) maintains gut homeostasis but can also promote inflammatory bowel disease (IBD). The regulation of ILC3-dependent colitis remains to be elucidated. Here we show that Foxp3(+) regulatory T cells (Treg cells) prevented ILC3-mediated colitis in an IL-10-independent manner. Treg cells inhibited IL-23 and IL-1beta production from intestinal-resident CX3CR1(+) macrophages but not CD103(+) dendritic cells. Moreover, Treg cells restrained ILC3 production of IL-22 through suppression of CX3CR1(+) macrophage production of IL-23 and IL-1beta. This suppression was contact dependent and was mediated by latent activation gene-3 (LAG-3)-an immune checkpoint receptor-expressed on Treg cells. Engagement of LAG-3 on MHC class II drove profound immunosuppression of CX3CR1(+) tissue-resident macrophages. Our study reveals that the health of the intestinal mucosa is maintained by an axis driven by Treg cells communication with resident macrophages that withhold inflammatory stimuli required for ILC3 function.
in vivo neutrophil depletion
Ellis, G. T., et al (2015). "TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection" EMBO Rep 16(9): 1203-1218.
PubMed
Streptococcus pneumoniae coinfection is a major cause of influenza-associated mortality; however, the mechanisms underlying pathogenesis or protection remain unclear. Using a clinically relevant mouse model, we identify immune-mediated damage early during coinfection as a new mechanism causing susceptibility. Coinfected CCR2(-/-) mice lacking monocytes and monocyte-derived cells control bacterial invasion better, show reduced epithelial damage and are overall more resistant than wild-type controls. In influenza-infected wild-type lungs, monocytes and monocyte-derived cells are the major cell populations expressing the apoptosis-inducing ligand TRAIL. Accordingly, anti-TRAIL treatment reduces bacterial load and protects against coinfection if administered during viral infection, but not following bacterial exposure. Post-influenza bacterial outgrowth induces a strong proinflammatory cytokine response and massive inflammatory cell infiltrate. Depletion of neutrophils or blockade of TNF-alpha facilitate bacterial outgrowth, leading to increased mortality, demonstrating that these factors aid bacterial control. We conclude that inflammatory monocytes recruited early, during the viral phase of coinfection, induce TRAIL-mediated lung damage, which facilitates bacterial invasion, while TNF-alpha and neutrophil responses help control subsequent bacterial outgrowth. We thus identify novel determinants of protection versus pathology in influenza-Streptococcus pneumoniae coinfection.
in vivo TIM-3 neutralization
Kurtulus, S., et al (2015). "TIGIT predominantly regulates the immune response via regulatory T cells" J Clin Invest. doi : 10.1172/JCI81187.
PubMed
Coinhibitory receptors are critical for the maintenance of immune homeostasis. Upregulation of these receptors on effector T cells terminates T cell responses, while their expression on Tregs promotes their suppressor function. Understanding the function of coinhibitory receptors in effector T cells and Tregs is crucial, as therapies that target coinhibitory receptors are currently at the forefront of treatment strategies for cancer and other chronic diseases. T cell Ig and ITIM domain (TIGIT) is a recently identified coinhibitory receptor that is found on the surface of a variety of lymphoid cells, and its role in immune regulation is just beginning to be elucidated. We examined TIGIT-mediated immune regulation in different murine cancer models and determined that TIGIT marks the most dysfunctional subset of CD8+ T cells in tumor tissue as well as tumor-tissue Tregs with a highly active and suppressive phenotype. We demonstrated that TIGIT signaling in Tregs directs their phenotype and that TIGIT primarily suppresses antitumor immunity via Tregs and not CD8+ T cells. Moreover, TIGIT+ Tregs upregulated expression of the coinhibitory receptor TIM-3 in tumor tissue, and TIM-3 and TIGIT synergized to suppress antitumor immune responses. Our findings provide mechanistic insight into how TIGIT regulates immune responses in chronic disease settings.
in vivo TIM-3 neutralization
in vivo blocking of PD-1/PD-L signaling
in vivo PD-L1 blockade
Flow Cytometry
Ngiow, S. F., et al (2015). "A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1" Cancer Res 75(18): 3800-3811.
PubMed
Despite successes, thus far, a significant proportion of the patients treated with anti-PD1 antibodies have failed to respond. We use mouse tumor models of anti-PD1 sensitivity and resistance and flow cytometry to assess tumor-infiltrating immune cells immediately after therapy. We demonstrate that the expression levels of T-cell PD1 (PD1(lo)), myeloid, and T-cell PDL1 (PDL1(hi)) in the tumor microenvironment inversely correlate and dictate the efficacy of anti-PD1 mAb and function of intratumor CD8(+) T cells. In sensitive tumors, we reveal a threshold for PD1 downregulation on tumor-infiltrating CD8(+) T cells below which the release of adaptive immune resistance is achieved. In contrast, PD1(hi) T cells in resistant tumors fail to be rescued by anti-PD1 therapy and remain dysfunctional unless intratumor PDL1(lo) immune cells are targeted. Intratumor Tregs are partly responsible for the development of anti-PD1-resistant tumors and PD1(hi) CD8(+) T cells. Our analyses provide a framework to interrogate intratumor CD8(+) T-cell PD1 and immune PDL1 levels and response in human cancer. Cancer Res; 75(18); 3800-11. (c)2015 AACR.
in vivo IFNγ neutralization
Flow Cytometry
Simons, D. M., et al (2013). "Autoreactive Th1 cells activate monocytes to support regional Th17 responses in inflammatory arthritis" J Immunol 190(7): 3134-3141.
PubMed
We have examined mechanisms underlying the formation of pathologic Th17 cells using a transgenic mouse model in which autoreactive CD4(+) T cells recognize influenza virus hemagglutinin (HA) as a ubiquitously expressed self-Ag and induce inflammatory arthritis. The lymph nodes of arthritic mice contain elevated numbers of inflammatory monocytes (iMO) with an enhanced capacity to promote CD4(+) Th17 cell differentiation, and a regional inflammatory response develops in the paw-draining lymph nodes by an IL-17-dependent mechanism. The activation of these Th17-trophic iMO precedes arthritis development and occurs in the context of an autoreactive CD4(+) Th1 cell response. Adoptive transfer of HA-specific CD4(+) T cells into nonarthritic mice expressing HA as a self-Ag similarly led to the formation of Th1 cells and of iMO that could support Th17 cell formation, and, notably, the accumulation of these iMO in the lymph nodes was blocked by IFN-gamma neutralization. These studies show that autoreactive CD4(+) Th1 cells directed to a systemically distributed self-Ag can promote the development of a regional Th17 cell inflammatory response by driving the recruitment of Th17-trophic iMO to the lymph nodes.
in vivo neutrophil depletion
Flow Cytometry
Bamboat, Z. M., et al (2010). "Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion" J Clin Invest 120(2): 559-569.
PubMed
TLRs are recognized as promoters of tissue damage, even in the absence of pathogens. TLR binding to damage-associated molecular patterns (DAMPs) released by injured host cells unleashes an inflammatory cascade that amplifies tissue destruction. However, whether TLRs possess the reciprocal ability to curtail the extent of sterile inflammation is uncertain. Here, we investigated this possibility in mice by studying the role of conventional DCs (cDCs) in liver ischemia/reperfusion (I/R) injury, a model of sterile inflammation. Targeted depletion of mouse cDCs increased liver injury after I/R, as assessed by serum alanine aminotransferase and histologic analysis. In vitro, we identified hepatocyte DNA as an endogenous ligand to TLR9 that promoted cDCs to secrete IL-10. In vivo, cDC production of IL-10 required TLR9 and reduced liver injury. In addition, we found that inflammatory monocytes recruited to the liver via chemokine receptor 2 were downstream targets of cDC IL-10. IL-10 from cDCs reduced production of TNF, IL-6, and ROS by inflammatory monocytes. Our results implicate inflammatory monocytes as mediators of liver I/R injury and reveal that cDCs respond to DAMPS during sterile inflammation, providing the host with protection from progressive tissue damage.
in vivo CTLA-4 neutralization
in vivo TIM-3 neutralization
in vivo blocking of PD-1/PD-L signaling
Mittal, D., et al (2014). "Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor" Cancer Res 74(14): 3652-3658.
PubMed
Adenosine targeting is an attractive new approach to cancer treatment, but no clinical study has yet examined adenosine inhibition in oncology despite the safe clinical profile of adenosine A2A receptor inhibitors (A2ARi) in Parkinson disease. Metastasis is the main cause of cancer-related deaths worldwide, and therefore we have studied experimental and spontaneous mouse models of melanoma and breast cancer metastasis to demonstrate the efficacy and mechanism of a combination of A2ARi in combination with anti-PD-1 monoclonal antibody (mAb). This combination significantly reduces metastatic burden and prolongs the life of mice compared with either monotherapy alone. Importantly, the combination was only effective when the tumor expressed high levels of CD73, suggesting a tumor biomarker that at a minimum could be used to stratify patients that might receive this combination. The mechanism of the combination therapy was critically dependent on NK cells and IFNgamma, and to a lesser extent, CD8(+) T cells and the effector molecule, perforin. Overall, these results provide a strong rationale to use A2ARi with anti-PD-1 mAb for the treatment of minimal residual and metastatic disease.
Product Citations
-
-
Cardiovascular biology
Neuronal Serpina3n is an endogenous protector against blood brain barrier damage following cerebral ischemic stroke.
In Journal of Cerebral Blood Flow & Metabolism on 1 February 2023 by Li, F., Zhang, Y., et al.
PubMed
Ischemic stroke results in blood-brain barrier (BBB) disruption, during which the reciprocal interaction between ischemic neurons and components of the BBB appears to play a critical role. However, the underlying mechanisms for BBB protection remain largely unknown. In this study, we found that Serpina3n, a serine protease inhibitor, was significantly upregulated in the ischemic brain, predominantly in ischemic neurons from 6 hours to 3 days after stroke. Using neuron-specific adeno-associated virus (AAV), intranasal delivery of recombinant protein, and immune-deficient Rag1-/- mice, we demonstrated that Serpina3n attenuated BBB disruption and immune cell infiltration following stroke by inhibiting the activity of granzyme B (GZMB) and neutrophil elastase (NE) secreted by T cells and neutrophils. Furthermore, we found that intranasal delivery of rSerpina3n significantly attenuated the neurologic deficits after stroke. In conclusion, Serpina3n is a novel ischemic neuron-derived proteinase inhibitor that counterbalances BBB disruption induced by peripheral T cell and neutrophil infiltration after ischemic stroke. These findings reveal a novel endogenous protective mechanism against BBB damage with Serpina3n being a potential therapeutic target in ischemic stroke.
-
-
TNF Superfamily Member 14 Drives Post-Influenza Depletion of Alveolar Macrophages Enabling Secondary Pneumococcal Pneumonia.
In J Clin Invest on 18 November 2025 by Malainou, C., Peteranderl, C., et al.
PubMed
Secondary bacterial infection, often caused by Streptococcus pneumoniae (Spn), is one of the most frequent and severe complications of influenza A virus (IAV)-induced pneumonia. Phenotyping of the pulmonary immune cell landscape after IAV infection revealed a substantial depletion of the tissue-resident alveolar macrophage (TR-AM) population at day 7, which was associated with increased susceptibility to Spn outgrowth. To elucidate the molecular mechanisms underlying TR-AM depletion, and to define putative targets for treatment, we combined single-cell transcriptomics and cell-specific PCR profiling in an unbiased manner, using in vivo models of IAV infection and IAV/Spn co-infection. The TNF superfamily 14 (TNFSF14) ligand-receptor axis was revealed as the driving force behind post-influenza TR-AM death during the early infection phase, enabling the transition to pneumococcal pneumonia, while intrapulmonary transfer of genetically modified TR-AMs and antibody-mediated neutralization of specific pathway components alleviated disease severity. With a mainly neutrophilic expression and a high abundance in the bronchoalveolar fluid (BALF) of patients with severe virus-induced ARDS, TNFSF14 emerged as a key determinant of virus-driven lung injury. Targeting the TNFSF14-mediated intercellular communication network in the virus-infected lung can, therefore, improve host defense, minimizing the risk of subsequent bacterial pneumonia, and ameliorating disease outcome.
-
-
Cancer Research
Cryoablation plus sintilimab and lenvatinib in advanced or metastatic intrahepatic cholangiocarcinoma: a phase 2 trial.
In Nat Cancer on 1 November 2025 by Gu, S., Luo, Q., et al.
PubMed
Treatment options for advanced or metastatic intrahepatic cholangiocarcinoma (ICC) are limited. In this single-arm, phase 2 trial (CASTLE-01, NCT05010668 ), 28 participants with advanced or metastatic ICC who have progressed after chemotherapy were treated with cryoablation, followed by anti-PD1 sintilimab (200 mg every 3 weeks) plus lenvatinib (8-12 mg per day) 2 weeks later. The objective response rate assessed by Response Evaluation Criteria in Solid Tumors version 1.1 was 75.0% (95% confidence interval (CI): 59-91%), meeting the prespecified primary endpoint. Secondary endpoints of disease control rate, median progression-free survival and overall survival were respectively 100% (95% CI: 100-100%), 16.8 months (95% CI: 11.5-not reached (NR)) and 25.4 months (95% CI: 13.3-NR). Treatment was well tolerated. Post hoc multiomics analysis of paired pretreatment and on-treatment tumor biopsies suggested that cryoablation increased the tumor immunogenicity and dendritic cell activation, followed by triggering continuous replenishment of intratumoral CD8+PD1hi effectors from peripheral blood. The addition of lenvatinib transitioned endothelial cells into inflamed venules to boost lymphocyte influx and targeted tumor stroma to promote CD8+PD1hi effectors penetrating into tumor cell nests. Therefore, cryoablation combined with sintilimab plus lenvatinib represents a promising approach for the treatment of advanced or metastatic ICC. These findings also support the notion that cryoablation may trigger abscopal antitumor immunity in ICC when combined with lenvatinib and PD1 blockade. ClinicalTrials.gov registration: NCT05010668 .
-
-
-
Cancer Research
A pan-cancer single-cell analysis reveals the effect of PD-1 blockades on tumor angiogenesis by inhibiting the endothelial CXCL12-CXCR4 axis.
In Cancer Immunol Immunother on 9 October 2025 by Muhetarijiang, M., Zhu, P., et al.
PubMed
Immune checkpoint inhibitors (ICIs), particularly programmed cell death protein 1 (PD-1) blockades, have redefined oncology in the last decade. Previous studies on PD-1 blockades mostly concentrate on their interactions with immune cells. This study aims to investigate how PD-1 blockades affect endothelial cell (EC) heterogeneity in the tumor microenvironment (TME) and to explore potential targets for enhancing the anti-tumor effects of PD-1 blockades. Here, we established a pan-cancer EC atlas from the public database and revealed that PD-1 blockades repress the angiogenic population in ECs and inhibit the CXCL12-CXCR4 signaling derived from ECs. Using a murine tumor model built with Lewis Lung Carcinoma cell line, we further validated our findings that a PD-1 blockade, as well as a CXCR4 antagonist AMD3100, inhibited EC population in tumors and their CXCL12 expression. In addition, the combo therapy of the PD-1 blockade and AMD3100 showed superior anti-tumor effects to monotherapy. Moreover, we predicted MYC to be the potential regulator through which PD-1 blockades affect ECs. Together, our results suggest that PD-1 blockades have an anti-angiogenic effect besides boosting T cell immunity, and the CXCL12/CXCR4 pathway is a potential target for enhancing the effectiveness of PD-1 blockades.
-
-
-
Cancer Research
-
Neuroscience
Cancer-induced nerve injury promotes resistance to anti-PD-1 therapy.
In Nature on 1 October 2025 by Baruch, E. N., Gleber-Netto, F. O., et al.
PubMed
Perineural invasion (PNI) is a well-established factor of poor prognosis in multiple cancer types1, yet its mechanism remains unclear. Here we provide clinical and mechanistic insights into the role of PNI and cancer-induced nerve injury (CINI) in resistance to anti-PD-1 therapy. Our study demonstrates that PNI and CINI of tumour-associated nerves are associated with poor response to anti-PD-1 therapy among patients with cutaneous squamous cell carcinoma, melanoma and gastric cancer. Electron microscopy and electrical conduction analyses reveal that cancer cells degrade the nerve fibre myelin sheets. The injured neurons respond by autonomously initiating IL-6- and type I interferon-mediated inflammation to promote nerve healing and regeneration. As the tumour grows, the CINI burden increases, and its associated inflammation becomes chronic and skews the general immune tone within the tumour microenvironment into a suppressive and exhaustive state. The CINI-driven anti-PD-1 resistance can be reversed by targeting multiple steps in the CINI signalling process: denervating the tumour, conditional knockout of the transcription factor mediating the injury signal within neurons (Atf3), knockout of interferon-α receptor signalling (Ifnar1-/-) or by combining anti-PD-1 and anti-IL-6-receptor blockade. Our findings demonstrate the direct immunoregulatory roles of CINI and its therapeutic potential.
-
-
Neutralization of Receptor activator of nuclear factor-κB ligand reduces fibrosis and promotes osteoblast differentiation in a mouse model of fibrous dysplasia driven by somatic expression of GnasR201H.
In JBMR Plus on 1 October 2025 by Ormsby, R. T., Zhang, Y., et al.
PubMed
Fibrous dysplasia (FD) is a rare disorder caused by somatic activating mutations in GNAS, encoding the alpha subunit of the Gs protein. Activating GNAS mutations result in focal expansile bone lesions, which cause pain, deformity, and increased risk of fracture. Somatic mosaicism in FD leads to both GNAS mutant and genetically WT osteoprogenitor cells, which jointly contribute to the formation of fibrotic lesions within the bone. Additionally, these lesions contain numerous osteoclasts formed in response to robust lesional expression of RANKL. Neutralizing antibody to RANKL is effective in reducing lesion growth in patients with FD and in preclinical models. To determine the effect of RANKL neutralization specifically on mutant cells early after onset of FD, we used a murine model of C57BL/6 Sox9CreERT;Gnas(R201H)fl/+;Rosa26LSL-tdTomato mice, which recapitulates the somatic mosaicism of FD bone lesions and in which mutant cells are lineage traced. Analysis of Gnas(R201H)fl/+ mice showed a diffuse accumulation of SMA+ early osteoblastic cells, with contribution from both tdTomato+ mutant and tdTomato- WT populations. Anti-RANKL treatment of Gnas(R201H)fl/+ mice inhibited osteoclast formation and substantially reduced fibrosis, detected by Masson's trichrome staining within the proximal metaphysis of the femur and the femoral head. Treatment with anti-RANKL decreased the accumulation of both mutant and WT SMA+ cells, accompanied by an increased number of mutant cells expressing the mature osteoblast marker osteocalcin, and an increase in overall osteoblast density. To elucidate the role of RANKL expression by mutant cells in the formation of FD lesions, we generated Sox9CreERT;Gnas(R201H)fl/+;Rosa26LSL-tdTomato;Ranklfl/fl mice. Deletion of Rankl in Gnas(R201H)fl/+ mutant cells did not prevent fibrosis in this model. The results suggest that while anti-RANKL treatment promotes osteoprogenitor differentiation to reduce fibrosis, the loss of RANKL expression from GNAS mutant cells alone is not sufficient to reverse the pathology of FD bone lesions.
-
-
Immunology and Microbiology
Identification and validation of intratumoral microbiome associated with sensitization to immune checkpoint inhibitors.
In Cell Rep Med on 16 September 2025 by Chen, J., Gao, Y., et al.
PubMed
As a part of the commensal microbiome, the regulatory role of the intratumoral microbiome in tumor immunity is gradually revealed. However, the relationship between the intratumoral microbiome and the efficacy of immune checkpoint inhibitors (ICIs) clinical treatment remains unclear. Here, we collect RNA sequencing (RNA-seq) data and clinical information from publicly available ICIs therapy cohorts. By developing an improved bioinformatics pipeline to identify the intratumoral microbiome and performing a comprehensive association analysis, we find that the intratumoral microbiome is associated with response to ICIs and characteristics of the tumor microenvironment (TME). In vivo experiments demonstrate that intratumoral injection of Burkholderia cepacia, Priestia megaterium, or Corynebacterium kroppenstedtii, which were selected from our analysis results, would synergize with anti-PD-1 therapy to inhibit tumor growth and enhance antitumor immunity. Our findings highlight the essential role of the intratumoral microbiome in the clinical effectiveness differences of ICIs, suggesting its potential in future ICIs combination therapy.
-
-
-
Immunology and Microbiology
-
Cell Biology
-
Cancer Research
CAD hijacks STING to impair antitumor immunity and radiotherapy efficacy of colorectal cancer.
In Cell Death Dis on 23 August 2025 by Cai, Z., Cheng, Z., et al.
PubMed
Radiotherapy (RT)-elicited antitumor immunity serves as a pivotal mechanism in RT-mediated tumor control. The strategic integration of RT with immunotherapies, particularly immune checkpoint blockade (ICB), is revolutionizing cancer therapeutics, demonstrating remarkable clinical potential. In this context, identifying molecular targets to potentiate radioimmunotherapy (RIT) efficacy represents a critical research priority. Emerging as a central immunomodulatory axis, the cGAS/STING signaling pathway bridges DNA damage response with antitumor immunity, positioning itself as a prime therapeutic target for radiation sensitization. Our study unveils caspase-activated DNase (CAD) as a previously unrecognized suppressor of cGAS/STING signaling that governs radiosensitivity in colorectal cancer (CRC). CAD physically blocks STING dimerization and cGAMP binding through a nuclease-independent function, thereby compromising RT-induced STING activation and subsequent type I interferon (IFN-I) production. Functional analyses demonstrated that CAD ablation potentiates CD8+ T cell infiltration/activation within the tumor microenvironment and synergizes with anti-PD-1 immunotherapy upon radiation. Translational validation revealed clinical correlations between CAD overexpression in CRC specimens and suboptimal radiotherapy responses coupled with diminished intratumoral CD8+ T cell infiltration. Collectively, our findings establish CAD as a novel rheostat of cGAS-STING signaling and propose CAD inhibition as a promising combinatorial strategy to enhance RT and RIT efficacy in CRC.
-
-
-
Immunology and Microbiology
-
Cancer Research
TIM3+ breast cancer cells license immune evasion during micrometastasis outbreak.
In Cancer Cell on 11 August 2025 by Rozalén, C., Sangrador, I., et al.
PubMed
In metastasis, the dynamics of tumor-immune interactions during micrometastasis remain unclear. Identifying the vulnerabilities of micrometastases before outbreaking into macrometastases can reveal therapeutic opportunities for metastasis. Here, we report a function of T cell immunoglobulin and mucin domain 3 (TIM3) in tumor cells during micrometastasis using breast cancer (BC) metastasis mouse models. TIM3 is highly upregulated in micrometastases, promoting survival, stemness, and immune escape. TIM3+ tumor cells are specifically selected during early seeding of micrometastasis. Mechanistically, TIM3 increases β-catenin/interleukin-1β (IL-1β) signaling, leading to stemness and immune-evasion by inducing immunosuppressive γδ T cells and reducing CD8 T cells during micrometastasis. Clinical data confirm increased TIM3+ tumor cells in BC metastasis and TIM3+ tumor cells as a biomarker of poor outcome in BC patients. (Neo)adjuvant TIM3 blockade reduces the metastatic seeding and incidence in preclinical models. These findings unveil a specific mechanism of micrometastasis immune-evasion and the potential use of TIM3 blockade for subclinical metastasis.
-
-
-
Immunology and Microbiology
-
Cancer Research
Thermosensitive Hydrogel Sustaining the Release of Lymph-Draining Oligonucleotide Adjuvant Polyplex Micelles Improves Systemic Cancer Immunotherapy.
In ACS Nano on 17 June 2025 by Lucas, S. N., Archer, P. A., et al.
PubMed
Immune checkpoint blockade (ICB) immunotherapies are a powerful tool in the clinical management of cancer, but response rates to ICB remain limited, and treatment-related toxicities can be significant. Therapeutic efficacy of ICB can be enhanced by delivering synergistic immunomodulators to tumor-draining lymph nodes (TdLNs). However, achieving sustained release of small molecule immunomodulators into the lymphatics and TdLNs remains challenging. To address this limitation, a sustained release system for delivering an oligonucleotide adjuvant to lymph nodes (LNs) was developed. CpG oligonucleotide was complexed with a redox-responsive cationic polymer and mixed with F127-g-Gelatin to generate a thermosensitive hydrogel that releases lymph-draining polyplex micelles in situ. This CpG/BPEI-SS-/F127-g-Gelatin (CpG-HG) system enhanced the quantity and duration of CpG delivery to TdLNs following locoregional administration compared with free drug and enabled targeted, potent, and prolonged immunomodulation within TdLNs from a single administration. This augmented, localized immune response synergized with systemic ICB treatment, both markedly amplifying the systemic circulating CD8+ T cell response and improving antitumor therapeutic efficacy while enabling ICB dose reduction. These results highlight the potential for this drug delivery system as an adjunct to existing clinical ICB protocols to improve patient outcomes.
-
-
Type I interferon protects against bone loss in periodontitis by mitigating an interleukin (IL)-17-neutrophil axis.
In Life Sci on 15 June 2025 by Zhang, J., Ding, Q., et al.
PubMed
Type I interferons (IFNs-I), a group of pleiotropic cytokines, critically modulate host response in various inflammatory diseases. However, the role of the IFN-I pathway in periodontitis remains largely unknown. In this report, we describe that the IFN-β levels in the gingival crevicular fluid of human subjects were negatively associated with periodontitis and clinical gingival inflammation. Disruption of IFN-I signaling worsened alveolar bone resorption in a ligature-induced periodontitis murine model. Deficiency of the IFN-I pathway resulted in an exaggerated inflammatory response in myeloid cells and drastically increased the interleukin-17 (IL-17)-mediated neutrophil recruitment in the gingiva. We further identified that the myeloid lineage-specific IFN-I response was essential in safeguarding against periodontal inflammation by suppressing the IL-17-producing γδ T cells in gingiva. IFN-I signaling also directly repressed osteoclastogenesis in monocytes, which are precursor cells for osteoclasts. Therefore, our findings demonstrate that an integral myeloid-specific IFN-I pathway protects against bone loss by keeping the IL-17-neutrophil axis in check and directly inhibiting osteoclast formation in periodontitis.
-
-
Immunology and Microbiology
APOE protects against severe infection with Mycobacterium tuberculosis by restraining production of neutrophil extracellular traps.
In PLoS Pathog on 1 June 2025 by Liu, D., Mai, D., et al.
PubMed
Mice lacking apolipoprotein E (APOE, Apoe-/- mice) on a high cholesterol (HC) diet are highly susceptible to infection with Mycobacterium tuberculosis (Mtb) but the underlying immune dysregulation has been unclear. While neutrophils are often the predominant cell type in the lungs of humans with severe tuberculosis (TB), they are relatively scarce in the lungs of some strains of mice that are used to study the disease. The neutrophil levels in the lungs of Mtb-infected Apoe-/- HC mice are very high, and thus studies in this model offer the opportunity to examine the role of specific neutrophil functions in the pathology of severe TB. We determined that depleting neutrophils, depleting plasmacytoid dendritic cells (pDCs), or blocking type I interferon signaling improved the outcome of TB in Apoe-/- HC mice. We also demonstrated that blocking the activation of peptidylarginine deiminase 4 (PAD4), an enzyme critical to NET formation, leads to fewer NETs in the lungs and dramatically improves the outcome of TB in Apoe-/- HC mice without affecting the number of neutrophils in the lung. We found that the transcriptional profile of neutrophils in Mtb-infected Apoe-/- HC mice is biased towards a state that resembles the "N2" phenotype that has been defined in cancer models and has been implicated in matrix degradation and tissue destruction. Our observations strongly suggest that the state of the neutrophil when it encounters the Mtb-infected lung is one of the main drivers of severe disease and implies that targeted interventions that alter specific states or functions, such as the production of NETs, may improve outcome while preserving sufficient capacity for host-defense.
-
-
-
In vivo experiments
-
Rattus norvegicus (Rat)
Differential regulation of fetal bone marrow and liver hematopoiesis by yolk-sac-derived myeloid cells.
In Nat Commun on 14 May 2025 by Weinhaus, B., Homan, S., et al.
PubMed
Fetal hematopoiesis takes place in the liver before colonizing the bone marrow where it will persist for life. This colonization is thought to be mediated by specification of a microenvironment that selectively recruits hematopoietic cells to the nascent bone marrow. The identity and mechanisms regulating the specification of this colonization niche are unclear. Here we identify a VCAM1+ sinusoidal colonization niche in the diaphysis that regulates neutrophil and hematopoietic stem cell colonization of the bone marrow. Using confocal microscopy, we find that colonizing hematopoietic stem and progenitor cells (HSPC) and myeloid cells selectively localize to a subset of VCAM1+ sinusoids in the center of the diaphysis. Vcam1 deletion in endothelial cells impairs hematopoietic colonization while depletion of yolk-sac-derived osteoclasts disrupts VCAM1+ expression, and impairs neutrophil and HSPC colonization to the bone marrow. Depletion of yolk-sac-derived myeloid cells increases fetal liver hematopoietic stem cell numbers, function and erythropoiesis independent of osteoclast activity. Thus, the yolk sac produces myeloid cells that have opposite roles in fetal hematopoiesis: while yolk-sac derived myeloid cells in the bone marrow promote hematopoietic colonization by specifying a VCAM1+ colonization niche, a different subset of yolk-sac-derived myeloid cells inhibits HSC in the fetal liver.
-
-
-
Cancer Research
-
Immunology and Microbiology
CRISPR screens in the context of immune selection identifyCHD1andMAP3K7as mediators of cancer immunotherapy resistance
In bioRxiv on 18 April 2025 by Watterson, A., Picco, G., et al.
-
-
-
Cancer Research
-
Immunology and Microbiology
Pseudomonas aeruginosa enhances anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8+ T cells.
In Front Immunol on 7 April 2025 by Chen, L., Ruan, G., et al.
PubMed
Immune checkpoint therapy for colorectal cancer (CRC) has been found to be unsatisfactory for clinical treatment. Fecal microbiota transplantation (FMT) has been shown to remodel the intestinal flora, which may improve the therapeutic effect of αPD-1. Further exploration of key genera that can sensitize cells to αPD-1 for CRC treatment and preliminary exploration of immunological mechanisms may provide effective guidance for the clinical treatment of CRC.
-
-
-
Immunology and Microbiology
Identification of a group of 9-amino-acridines that selectively downregulate regulatory T cell functions through FoxP3.
In iScience on 21 March 2025 by Wei, Q., Foyn, H., et al.
PubMed
FoxP3+ regulatory T cells (Tregs) are responsible for immune homeostasis by suppressing excessive anti-self-immunity. Tregs facilitate tumor growth by inhibiting anti-tumor immunity. Here, we explored the targeting of FoxP3 as a basis for new immunotherapies. In a high-throughput phenotypic screening of a drug repurposing library using human primary T cells, we identified quinacrine as a FoxP3 downregulator. In silico searches based on the structure of quinacrine, testing of sub-libraries of analogs in vitro, and validation identified a subset of 9-amino-acridines that selectively abrogated Treg suppressive functions. Mechanistically, these acridines interfered with the DNA-binding activity of FoxP3 and inhibited FoxP3-regulated downstream gene regulation. Release from Treg suppression by 9-amino-acridines increased anti-tumor immune responses both in cancer patient samples and in mice in a syngeneic tumor model. Our study highlights the feasibility of screening for small molecular inhibitors of FoxP3 as an approach to pursuing Treg-based immunotherapy.
-
-
-
Immunology and Microbiology
Blocking WNT7A Enhances MHC-I Antigen Presentation and Enhances the Effectiveness of Immune Checkpoint Blockade Therapy.
In Cancer Immunol Res on 4 March 2025 by Sun, J., Wang, P., et al.
PubMed
The limited infiltration of CD8+ T cells in tumors hampers the effectiveness of T cell-based immunotherapy, yet the mechanisms that limit tumor infiltration by CD8+ T cells remain unclear. Through bulk RNA sequencing of human tumors, we identified a strong correlation between WNT7A expression and reduced CD8+ T-cell infiltration. Further investigation demonstrated that inhibiting WNT7A substantially enhanced MHC-I expression on tumor cells. Mechanistically, WNT7A inhibition inactivated the Wnt/β-catenin signaling pathway and thus resulted in reduced physical interaction between β-catenin and p65 in the cytoplasm, which increased the nuclear translocation of p65 and activated the NF-κB pathway, ultimately promoting the transcription of genes encoding MHC-I molecules. We found that our lead compound, 1365-0109, disrupted the protein-protein interaction between WNT7A and its receptor FZD5, resulting in the upregulation of MHC-I expression. In murine tumor models, both genetic and pharmaceutical suppression of WNT7A led to increased MHC-I levels on tumor cells, and consequently enhanced the infiltration and functionality of CD8+ T cells, which bolstered antitumor immunity and improved the effectiveness of immune checkpoint blockade therapy. These findings have elucidated the intrinsic mechanisms of WNT7A-induced immune suppression, suggesting that therapeutic interventions targeting WNT7A hold promise for enhancing the efficacy of immunotherapy.
-
-
-
Cancer Research
Metastatic tumor growth in steatotic liver is promoted by HAS2-mediated fibrotic tumor microenvironment.
In J Clin Invest on 13 February 2025 by Yang, Y. M., Kim, J., et al.
PubMed
Steatotic liver enhances liver metastasis of colorectal cancer (CRC), but this process is not fully understood. Steatotic liver induced by a high-fat diet increases cancer-associated fibroblast (CAF) infiltration and collagen and hyaluronic acid (HA) production. We investigated the role of HA synthase 2 (HAS2) in the fibrotic tumor microenvironment in steatotic liver using Has2ΔHSC mice, in which Has2 is deleted from hepatic stellate cells. Has2ΔHSC mice had reduced steatotic liver-associated metastatic tumor growth of MC38 CRC cells, collagen and HA deposition, and CAF and M2 macrophage infiltration. We found that low-molecular weight HA activates Yes-associated protein (YAP) in cancer cells, which then releases connective tissue growth factor to further activate CAFs for HAS2 expression. Single-cell analyses revealed a link between CAF-derived HAS2 and M2 macrophages and CRC cells through CD44; these cells were associated with exhausted CD8+ T cells via programmed death-ligand 1 and programmed cell death protein 1 (PD-1). HA synthesis inhibitors reduced steatotic liver-associated metastasis of CRC, YAP expression, and CAF and M2 macrophage infiltration, and improved response to anti-PD-1 antibody. In conclusion, steatotic liver modulates a fibrotic tumor microenvironment to enhance metastatic cancer activity through a bidirectional regulation between CAFs and metastatic tumors, enhancing the metastatic potential of CRC in the liver.
-
-
-
Cancer Research
-
Immunology and Microbiology
Harnessing macrophage-drug conjugates for allogeneic cell-based therapy of solid tumors via the TRAIN mechanism.
In Nat Commun on 4 February 2025 by Taciak, B., Bialasek, M., et al.
PubMed
Treatment of solid tumors remains challenging and therapeutic strategies require continuous development. Tumor-infiltrating macrophages play a pivotal role in tumor dynamics. Here, we present a Macrophage-Drug Conjugate (MDC) platform technology that enables loading macrophages with ferritin-drug complexes. We first show that macrophages actively take up human heavy chain ferritin (HFt) in vitro via macrophage scavenger receptor 1 (MSR1). We further manifest that drug-loaded macrophages transfer ferritin to adjacent cancer cells through a process termed 'TRAnsfer of Iron-binding protein' (TRAIN). The TRAIN process requires direct cell-to-cell contact and an immune synapse-like structure. At last, MDCs with various anti-cancer drugs are formulated with their safety and anti-tumor efficacy validated in multiple syngeneic mice and orthotopic human tumor models via different routes of administration. Importantly, MDCs can be prepared in advance and used as thawed products, supporting their clinical applicability. This MDC approach thus represents a promising advancement in the therapeutic landscape for solid tumors.
-
-
-
Cancer Research
-
Endocrinology and Physiology
-
Immunology and Microbiology
Caerin 1.1/1.9-mediated antitumor immunity depends on IFNAR-Stat1 signalling of tumour infiltrating macrophage by autocrine IFNα and is enhanced by CD47 blockade.
In Sci Rep on 30 January 2025 by Li, J., Luo, Y., et al.
PubMed
Previously, we demonstrated that natural host-defence peptide caerin 1.1/caerin 1.9 (F1/F3) increases the efficacy of anti-PD-1 and therapeutic vaccine, in a HPV16 + TC-1 tumour model, but the anti-tumor mechanism of F1/F3 is still unclear. In this study, we explored the impact of F1/F3 on the tumor microenvironment in a transplanted B16 melanoma model, and further investigated the mechanism of action of F1/F3 using monoclonal antibodies to deplete relevant cells, gene knockout mice and flow cytometry. We show that F1/F3 is able to inhibit the growth of melanoma B16 tumour cells both in vitro and in vivo. Depletion of macrophages, blockade of IFNα receptor, and Stat1 inhibition each abolishes F1/F3-mediated antitumor responses. Subsequent analysis reveals that F1/F3 increases the tumour infiltration of inflammatory macrophages, upregulates the level of IFNα receptor, and promotes the secretion of IFNα by macrophages. Interestingly, F1/F3 upregulates CD47 level on tumour cells; and blocking CD47 increases F1/F3-mediated antitumor responses. Furthermore, F1/F3 intratumor injection, CD47 blockade, and therapeutic vaccination significantly increases the survival time of B16 tumour-bearing mice. These results indicate that F1/F3 may be effective to improve the efficacy of ICB and therapeutic vaccine-based immunotherapy for human epithelial cancers and warrants consideration for clinical trials.
-