InVivoPlus anti-mouse Ly6G
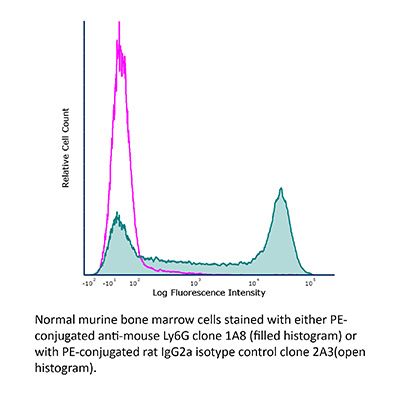
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | EL4J cells transfected with Ly6G |
| Reported Applications |
in vivo neutrophil depletion in vivo MDSC depletion Immunofluorescence Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Purification | Protein G |
| RRID | AB_1107721 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Deng, L., et al (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695.
PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Moynihan, K. D., et al (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200.
PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
-
Conde, P., et al (2015). "DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance" Immunity 42(6): 1143-1158.
PubMed
Tissue effector cells of the monocyte lineage can differentiate into different cell types with specific cell function depending on their environment. The phenotype, developmental requirements, and functional mechanisms of immune protective macrophages that mediate the induction of transplantation tolerance remain elusive. Here, we demonstrate that costimulatory blockade favored accumulation of DC-SIGN-expressing macrophages that inhibited CD8(+) T cell immunity and promoted CD4(+)Foxp3(+) Treg cell expansion in numbers. Mechanistically, that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling was required for production of immunoregulatory IL-10 associated with prolonged allograft survival. Deletion of DC-SIGN-expressing macrophages in vivo, interfering with their CSF1-dependent development, or preventing the DC-SIGN signaling pathway abrogated tolerance. Together, the results provide new insights into the tolerogenic effects of costimulatory blockade and identify DC-SIGN(+) suppressive macrophages as crucial mediators of immunological tolerance with the concomitant therapeutic implications in the clinic.
Product Citations
-
HIF-1α Promotes Macrophage Extracellular Trap Formation and Exacerbates Acute Lung Injury in Neonatal Sepsis.
In Biomedicines on 18 May 2026 by Zhang, H., Huang, W., et al.
PubMed
Background: Acute lung injury (ALI) is a major contributor to mortality in neonatal sepsis, yet the mechanisms underlying early lung damage remain incompletely understood. Although extracellular traps (ETs) have been implicated in inflammatory injury, the cellular origin and regulatory pathways of ET formation in neonatal sepsis remain unclear. This study aimed to determine the source of ETs and to investigate the role of hypoxia-inducible factor-1α (HIF-1α) in regulating macrophage extracellular traps (METs) formation and lung injury. Methods: Neonatal sepsis was induced in mice by intraperitoneal injection of cecal slurry. METs formation was assessed by immunofluorescence staining, Western blotting, and extracellular DNA quantification. Selective depletion of macrophages or neutrophils was performed to determine the cellular source of ETs. In vitro experiments were conducted using macrophages stimulated with lipopolysaccharide or phorbol 12-myristate 13-acetate. RNA sequencing analysis and pharmacological inhibition were used to examine the roles of HIF-1α, glycolysis, and enolase 2 (ENO2) in METs formation, lung injury, and survival outcomes. Results: We identify macrophages as a predominant source of ETs in the lung and demonstrate that METs contribute to lung injury in neonatal sepsis. Depletion of macrophages or pharmacological inhibition of METs formation markedly attenuated lung injury and improved survival in neonatal sepsis mice. Mechanistically, we suggest that HIF-1α promotes METs formation by driving glycolysis in macrophages. Furthermore, this process appears to involve the upregulation of key glycolytic enzymes, including ENO2, potentially facilitating METs release. In turn, METs are implicated in enhancing macrophage inflammatory activation, which could exacerbate lung injury. Importantly, pharmacological targeting of HIF-1α pathways reduces METs formation, attenuates lung inflammation, and improves survival outcomes. Conclusions: These findings suggest a role for HIF-1α in regulating METs formation and support that targeting this pathway could represent a potential therapeutic strategy for neonatal sepsis-associated acute lung injury.
-
Neutrophil depletion at the early stage of Japanese encephalitis virus infection affects CD8+ T cell infiltration into the mouse brain and causes severe encephalitis.
In Front Immunol on 6 February 2026 by Soni, R., Jena, P., et al.
PubMed
Neutrophils have been reported to have protective and detrimental functions in viral infections. However, the role of neutrophils remains unexplored in Japanese encephalitis virus (JEV) infection. In this study, we elucidated the dynamics of neutrophils and their influence on immune cell recruitment in subclinical and severe encephalitis in mouse models. Further, we depleted neutrophils from 3-4 week-old C57BL/6 mice using mAb1A8 (anti-Ly6G) antibody and studied their association with inflammation, viral replication, immune cell infiltration and disease outcome. We observed that an increase in JEV replication is associated with increased infiltration of neutrophils in the spleen and brain. Further studies confirmed that depletion of neutrophils at an early stage of JEV infection reduced CD8 abundance in the infected brain and accelerated death in mice. We also observed that inhibition of the CXCL12-CXCR4 signalling axis by antagonist AMD3100 reduced CD8 abundance in the brain and augmented inflammasome activation, leading to fatal encephalitis. Reduced CXCR4 levels in the spleen and blood of CD8+T cells correlated with enhanced Granzyme B level, indicating CD8 cells differentiated more into effector phenotypes in neutrophil-depleted mice. Furthermore, CD8 depletion delayed the death of mice infected with a sublethal strain compared to neutrophil-depleted mice, suggesting that neutrophils play a vital role in the early restriction of viral replication, whereas CD8 is essential later in clearing the virus. Taken together, our study sheds new light on the role of neutrophils in the pathogenic mechanisms of JEV encephalitis and highlights the importance of neutrophils and CD8 cells associated with disease outcomes.
-
TNF superfamily member 14 drives post-influenza depletion of alveolar macrophages, enabling secondary pneumococcal pneumonia.
In J Clin Invest on 16 January 2026 by Malainou, C., Peteranderl, C., et al.
PubMed
Secondary bacterial infection, often caused by Streptococcus pneumoniae, is one of the most frequent and severe complications of influenza A virus-induced (IAV-induced) pneumonia. Phenotyping of the pulmonary immune cell landscape after IAV infection revealed a substantial depletion of the tissue-resident alveolar macrophage (TR-AM) population at day 7, which was associated with increased susceptibility to S. pneumoniae outgrowth. To elucidate the molecular mechanisms underlying TR-AM depletion, and to define putative targets for treatment, we combined single-cell transcriptomics and cell-specific PCR profiling in an unbiased manner, using in vivo models of IAV infection and IAV and S. pneumoniae coinfection. The TNF superfamily 14 (TNFSF14) ligand/receptor axis was revealed as the driving force behind post-influenza TR-AM death during the early infection phase, enabling the transition to pneumococcal pneumonia, whereas intrapulmonary transfer of genetically modified TR-AMs and antibody-mediated neutralization of specific pathway components alleviated disease severity. With mainly neutrophilic expression and high abundance in the bronchoalveolar fluid of patients with severe virus-induced acute respiratory distress syndrome, TNFSF14 emerged as a key determinant of virus-driven lung injury. Targeting the TNFSF14-mediated intercellular communication network in the virus-infected lung can, therefore, improve host defense, minimizing the risk of subsequent bacterial pneumonia and ameliorating the disease outcome.
-
Neutrophils, not macrophages, aid phage-mediated control of pulmonary Pseudomonas aeruginosa infection.
In Front Immunol on 5 December 2025 by Weissfuss, C., Hoffmann, K., et al.
PubMed
The increasing prevalence of multidrug-resistant (MDR) bacteria has reduced the effectiveness of standard antibiotics, prompting renewed interest in bacteriophage (phage) therapy as an alternative or adjunctive treatment. Phage therapy offers high specificity, self-amplification at infection sites, and minimal disruption to the gut microbiota. However, clinical implementation is challenging, due to the risk of phage resistance and uncertainties regarding optimal dosing and immune interactions.