InVivoMAb anti-mouse Ly6G
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
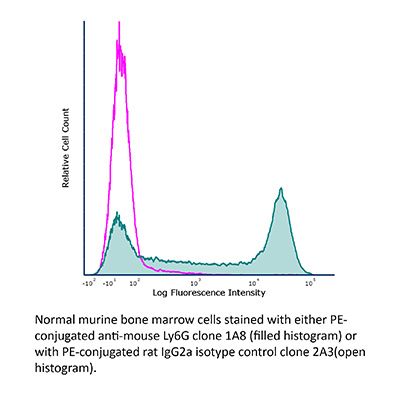
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | EL4J cells transfected with Ly6G |
| Reported Applications |
in vivo neutrophil depletion in vivo MDSC depletion Immunofluorescence Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Purification | Protein G |
| RRID | AB_1107721 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Deng, L., et al (2014). "Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice" J Clin Invest 124(2): 687-695.
PubMed
High-dose ionizing irradiation (IR) results in direct tumor cell death and augments tumor-specific immunity, which enhances tumor control both locally and distantly. Unfortunately, local relapses often occur following IR treatment, indicating that IR-induced responses are inadequate to maintain antitumor immunity. Therapeutic blockade of the T cell negative regulator programmed death-ligand 1 (PD-L1, also called B7-H1) can enhance T cell effector function when PD-L1 is expressed in chronically inflamed tissues and tumors. Here, we demonstrate that PD-L1 was upregulated in the tumor microenvironment after IR. Administration of anti-PD-L1 enhanced the efficacy of IR through a cytotoxic T cell-dependent mechanism. Concomitant with IR-mediated tumor regression, we observed that IR and anti-PD-L1 synergistically reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs), which suppress T cells and alter the tumor immune microenvironment. Furthermore, activation of cytotoxic T cells with combination therapy mediated the reduction of MDSCs in tumors through the cytotoxic actions of TNF. Our data provide evidence for a close interaction between IR, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and radiotherapy.
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Moynihan, K. D., et al (2016). "Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses" Nat Med. doi : 10.1038/nm.4200.
PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
-
Conde, P., et al (2015). "DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance" Immunity 42(6): 1143-1158.
PubMed
Tissue effector cells of the monocyte lineage can differentiate into different cell types with specific cell function depending on their environment. The phenotype, developmental requirements, and functional mechanisms of immune protective macrophages that mediate the induction of transplantation tolerance remain elusive. Here, we demonstrate that costimulatory blockade favored accumulation of DC-SIGN-expressing macrophages that inhibited CD8(+) T cell immunity and promoted CD4(+)Foxp3(+) Treg cell expansion in numbers. Mechanistically, that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling was required for production of immunoregulatory IL-10 associated with prolonged allograft survival. Deletion of DC-SIGN-expressing macrophages in vivo, interfering with their CSF1-dependent development, or preventing the DC-SIGN signaling pathway abrogated tolerance. Together, the results provide new insights into the tolerogenic effects of costimulatory blockade and identify DC-SIGN(+) suppressive macrophages as crucial mediators of immunological tolerance with the concomitant therapeutic implications in the clinic.
Product Citations
-
Angiotensin-converting enzyme overexpression in mouse neutrophils prevents Alzheimer's-like cognitive decline.
In Front Immunol on 6 May 2026 by Shibata, T., Bhat, S. A., et al.
PubMed
Angiotensin-converting enzyme (ACE), a dipeptidyl carboxypeptidase, is known to cleave amyloid-beta (Aβ), and its reduced activity has been linked to the progression of Alzheimer's disease (AD). Our research indicates that ACE is vital for myeloid cell functions. Using ACE10/10 recombinant mice, we demonstrated that overexpressing ACE in macrophages mitigates AD pathology in these mice. Given that neutrophils are the most abundant white blood cells, this study investigates whether ACE overexpression in neutrophils influences AD progression. We crossed NeuACE mice, which overexpress ACE in neutrophils, with 3xTg-AD mice to create AD-NeuACE mice. Behavioral changes and brain pathology were assessed through various behavioral mazes and histological assays. AD-NeuACE mice demonstrated improved cognitive functions and lower Aβ levels in the cortex and hippocampus compared to AD mice. In-vitro data indicate that ACE-overexpressing neutrophils are significantly more effective at phagocytosing and clearing Aβ fluorescence particles. This study suggests that overexpressing ACE in neutrophils could be a promising approach to managing AD-like phenotypes.
-
The hyaluronan receptor CD44 drives COVID-19 severity through its regulation of neutrophil migration.
In PLoS Pathog on 1 May 2026 by Hart, D. J., Uddin, M. J., et al.
PubMed
The novel respiratory disease COVID-19 caused by the coronavirus SARS-CoV-2 continues to be a public health emergency worldwide, and there is a need for more effective therapy for patients. The relationship between the extracellular matrix and the host immune response to infection is severely understudied. Deposition of the polysaccharide hyaluronan (HA) into the lungs is associated with more severe COVID-19 disease outcomes. HA is a major component of the extracellular matrix in connective tissues and is abundant in many parts of the body, including cartilage, skin, brain, and vitreous body. HA is a major component of the extracellular matrix in connective tissues and is abundant in many parts of the body, including cartilage, skin, brain, and vitreous body. Polymers consist of repeating units of N-acetylglucosamine and glucuronic acid and are synthesized by the three hyaluronan synthase (HAS) enzymes, HAS1-3. CD44 is the primary receptor for HA and is found on almost all immune cells in the lung. Known functions of CD44 include mediation of immune cell migration, activation, and differentiation. We hypothesized that increased HA deposition during COVID-19 increases CD44-mediated immune cell infiltration into lungs and results in more severe pathology. Here, we report that in mice infected with a mouse-adapted strain of SARS-CoV-2, treatment with a combination of two anti-CD44 monoclonal antibodies confers a significant survival benefit and reduces weight loss and clinical score of the mice on Day 4 post infection. We show that anti-CD44 treatment decreases many key cytokines and chemokines in the bronchoalveolar lavage fluid on Day 4. With flow cytometry, we show that anti-CD44 reduces the numbers of neutrophils in infected lungs. We also show through immunofluorescence that treatment with anti-CD44 antibodies reduces colocalization of HA and CD45 in lung sections, indicating that HA's interaction with immune cells contributes to pathology. Our findings demonstrate that disruption of HA-receptor interactions is a way to prevent inflammatory pathology in pulmonary infection.
-
Activated Platelet-Released Heat Shock Protein 90α Triggers Autophagy-Dependent Neutrophil Extracellular Trap Formation and Amplifies Sepsis.
In Adv Sci (Weinh) on 1 May 2026 by Wang, C., Leng, M., et al.
PubMed
Platelets are crucial to the development of thrombosis and coagulation abnormalities in sepsis, but the mechanisms by which they contribute to these pathological processes are not fully understood. Here, we identify a key role for platelet-released heat shock protein 90α (HSP90α) in driving neutrophil extracellular trap (NET) formation and supporting thromboinflammation during sepsis. Proteomic analysis of platelets from patients with sepsis showed a significant increase in HSP90α, which we traced back to trafficking pathways originating from megakaryocytes. When activated, platelets translocate HSP90α to their plasma membrane and release it into the extracellular space in both free and exosome-associated forms. Extracellular HSP90α acts as a damage-associated molecular pattern that binds to toll-like receptor 4 (TLR4) on neutrophils. This binding activates a downstream MyD88-Beclin 1 signaling pathway, triggering autophagy and leading to NET formation. Blocking extracellular HSP90α with a neutralizing monoclonal antibody significantly reduced NET formation both in vitro and in vivo, resulting in decreased sepsis-related thrombosis and inflammation. This platelet-HSP90α-TLR4-autophagy-NET pathway not only deepens our understanding of platelet-induced immunothrombosis but also suggests potential targets for therapies aimed at reducing coagulation problems and organ failure in septic patients.
-
Chemotherapy-induced activation of caspase-1 and IL-1α release by cancer cells remotely skews myelopoiesis to drive pro tumorigenic systemic neutrophil-dominant inflammation.
In Nat Commun on 20 April 2026 by Wong, S. Q. R., Hayashi, K., et al.
PubMed
While chemotherapy-induced tumor cell death is known to modulate the local immune landscape, its systemic impact on distant bone marrow-a site essential for immune cell maturation-remains underexplored. Here, we show that gemcitabine chemotherapy induces inflammatory caspase-1-dependent pyroptosis in epithelial cancer cells (epiCaspase-1). Despite its inflammatory nature, epiCaspase-1-mediated cell death is non-immunogenic. Clinically, elevated expression of an epiCaspase-1 gene signature correlates with worse patient outcomes. Mechanistically, epiCaspase-1 triggers the noncanonical release of IL-1α through NINJ1 lytic pores, remotely skewing bone marrow hematopoiesis towards granulocyte-monocyte progenitors and mature neutrophil output. This systemic reprogramming elevates the neutrophil-to-lymphocyte ratio (NLR) in both peripheral blood and the local tumor microenvironment. Pharmacological inhibition of caspase-1 and IL-1α disrupts this cascade, normalizes hematopoiesis, and recalibrates NLR by promoting intratumoral CD8+ T cell infiltration and activation, ultimately enhancing chemotherapeutic efficacy. These findings challenge the assumption that inflammatory pyroptosis is inherently immunogenic; instead, it can reshape systemic immune landscape towards a neutrophil-dominant inflammation in the chemotherapy context.