InVivoPlus anti-mouse IFNAR-1
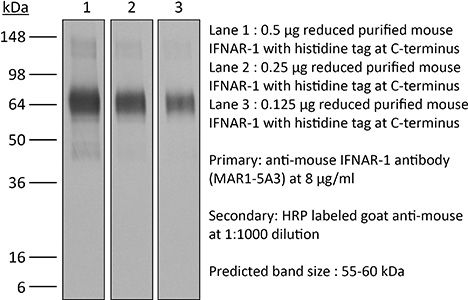
Product Description
Bio X Cell is pleased to also offer MAR1-5A3-CP056. This monoclonal antibody is a recombinant, chimeric version of the original MAR1-5A3 antibody. The variable domain sequences are identical to clone MAR1-5A3, but the constant region has been converted from mouse IgG1 to mouse IgG2a. MAR1-5A3-CP056 also contains Fc silencing mutations rendering it unable to bind to endogenous Fcγ receptors, similar to therapeutic anti-IFNAR-1 antibodies such as Anifrolumab. These mutations prevent Fc-effector functions like antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). The highly controlled sequence and lack of genetic drift in recombinant antibodies provide more reliable and reproducible results over hybridoma derived antibodies.
Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Extracellular domain of mouse IFNAR-1 |
| Reported Applications |
in vivo IFNAR-1 blockade in vitro IFNAR-1 blockade Western blot Flow Cytometry ELISA |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687723 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Liu, X., et al (2015). "CD47 blockade triggers T cell-mediated destruction of immunogenic tumors" Nat Med 21(10): 1209-1215.
PubMed
Macrophage phagocytosis of tumor cells mediated by CD47-specific blocking antibodies has been proposed to be the major effector mechanism in xenograft models. Here, using syngeneic immunocompetent mouse tumor models, we reveal that the therapeutic effects of CD47 blockade depend on dendritic cell but not macrophage cross-priming of T cell responses. The therapeutic effects of anti-CD47 antibody therapy were abrogated in T cell-deficient mice. In addition, the antitumor effects of CD47 blockade required expression of the cytosolic DNA sensor STING, but neither MyD88 nor TRIF, in CD11c(+) cells, suggesting that cytosolic sensing of DNA from tumor cells is enhanced by anti-CD47 treatment, further bridging the innate and adaptive responses. Notably, the timing of administration of standard chemotherapy markedly impacted the induction of antitumor T cell responses by CD47 blockade. Together, our findings indicate that CD47 blockade drives T cell-mediated elimination of immunogenic tumors.
-
Welten, S. P., et al (2015). "The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion" Elife 4. doi : 10.7554/eLife.07486.
PubMed
Signals delivered by costimulatory molecules are implicated in driving T cell expansion. The requirements for these signals, however, vary from dispensable to essential in different infections. We examined the underlying mechanisms of this differential T cell costimulation dependence and found that the viral context determined the dependence on CD28/B7-mediated costimulation for expansion of naive and memory CD8(+) T cells, indicating that the requirement for costimulatory signals is not imprinted. Notably, related to the high-level costimulatory molecule expression induced by lymphocytic choriomeningitis virus (LCMV), CD28/B7-mediated costimulation was dispensable for accumulation of LCMV-specific CD8(+) T cells because of redundancy with the costimulatory pathways induced by TNF receptor family members (i.e., CD27, OX40, and 4-1BB). Type I IFN signaling in viral-specific CD8(+) T cells is slightly redundant with costimulatory signals. These results highlight that pathogen-specific conditions differentially and uniquely dictate the utilization of costimulatory pathways allowing shaping of effector and memory antigen-specific CD8(+) T cell responses.
-
Beug, S. T., et al (2014). "Smac mimetics and innate immune stimuli synergize to promote tumor death" Nat Biotechnol 32(2): 182-190.
PubMed
Smac mimetic compounds (SMC), a class of drugs that sensitize cells to apoptosis by counteracting the activity of inhibitor of apoptosis (IAP) proteins, have proven safe in phase 1 clinical trials in cancer patients. However, because SMCs act by enabling transduction of pro-apoptotic signals, SMC monotherapy may be efficacious only in the subset of patients whose tumors produce large quantities of death-inducing proteins such as inflammatory cytokines. Therefore, we reasoned that SMCs would synergize with agents that stimulate a potent yet safe “cytokine storm.” Here we show that oncolytic viruses and adjuvants such as poly(I:C) and CpG induce bystander death of cancer cells treated with SMCs that is mediated by interferon beta (IFN-beta), tumor necrosis factor alpha (TNF-alpha) and/or TNF-related apoptosis-inducing ligand (TRAIL). This combinatorial treatment resulted in tumor regression and extended survival in two mouse models of cancer. As these and other adjuvants have been proven safe in clinical trials, it may be worthwhile to explore their clinical efficacy in combination with SMCs.
-
Macal, M., et al (2018). "Self-Renewal and Toll-like Receptor Signaling Sustain Exhausted Plasmacytoid Dendritic Cells during Chronic Viral Infection" Immunity 48(4): 730-744 e735.
PubMed
Although characterization of T cell exhaustion has unlocked powerful immunotherapies, the mechanisms sustaining adaptations of short-lived innate cells to chronic inflammatory settings remain unknown. During murine chronic viral infection, we found that concerted events in bone marrow and spleen mediated by type I interferon (IFN-I) and Toll-like receptor 7 (TLR7) maintained a pool of functionally exhausted plasmacytoid dendritic cells (pDCs). In the bone marrow, IFN-I compromised the number and the developmental capacity of pDC progenitors, which generated dysfunctional pDCs. Concurrently, exhausted pDCs in the periphery were maintained by self-renewal via IFN-I- and TLR7-induced proliferation of CD4(-) subsets. On the other hand, pDC functional loss was mediated by TLR7, leading to compromised IFN-I production and resistance to secondary infection. These findings unveil the mechanisms sustaining a self-perpetuating pool of functionally exhausted pDCs and provide a framework for deciphering long-term exhaustion of other short-lived innate cells during chronic inflammation.
Product Citations
-
PPP2R2A insufficiency enhances PD-L1 immune checkpoint blockade efficacy in lung cancer through cGAS-STING activation.
In J Clin Invest on 16 February 2026 by Qiu, Z., Song, N. J., et al.
PubMed
PP2A B55α, a regulatory subunit of protein phosphatase 2 (PP2A), is underexpressed in greater than 40% of non-small cell lung cancer (NSCLC) cases due to loss of heterozygosity of PPP2R2A, the gene encoding this protein. Given that low PPP2R2A expression correlates with poor prognosis, treating PPP2R2A-deficient NSCLC represents an unmet medical need. Here, we show that PPP2R2A knockdown or its heterozygosity (PPP2R2A+/-) increases cytosolic DNA, leading to cGAS-STING-type I IFN pathway activation. PPP2R2A deficiency results in elevated expression of immune checkpoint protein PD-L1 via GSK-3β- and STING-dependent mechanisms. PPP2R2A+/- cancer cells have enhanced sensitivity to PD-L1 blockade in a mouse model of lung cancer due to modulation of the tumor immune microenvironment, resulting in increased NK cells and reduced infiltration and function of Tregs. Consequently, PD-L1 antibody treatment increases CD8+ T infiltration and activity, especially in tumors with PPP2R2A heterozygosity. Furthermore, systemic or Treg-specific IFNAR1 blockade reduces the efficacy of PD-L1 blockade in PPP2R2A+/- tumors. Patients with NSCLC with a low PPP2R2A/PD-L1 ratio respond better to immune checkpoint blockade (ICB). These findings underscore the therapeutic potential of ICB in treating PPP2R2A-deficient NSCLC and suggest that PPP2R2A deficiency could serve as a biomarker for guiding ICB-based therapies.
-
Type I interferon signaling controls the early hematopoietic expansion in response to β-glucan.
In iScience on 16 May 2025 by Xu, Y., Lee, M. K. S., et al.
PubMed
Rapid hematopoietic adaptations are important for building and sustaining the biological response to β-glucan. The signals involved in these early events have not yet been fully explored. Given that type I interferons are produced in response to β-glucan and can profoundly impact hematopoietic stem cell (HSC) function, we hypothesized that this pathway may be involved in the early bone marrow response to β-glucan. In vivo administration of β-glucan led to local interferon-α production in the peritoneal cavity and bone marrow, upregulation of its receptor, IFNAR1, specifically on long-term hematopoietic stem cells (LT-HSCs), and broad expansion of downstream progenitor subpopulations. We demonstrate that intact type I interferon signaling is critical for β-glucan-mediated LT-HSC proliferation, mitochondrial activity, and glycolytic commitment. By determining that type I interferon signaling is important for LT-HSCs, which sit at the apex of the hematopoietic hierarchy, we uncover an important component of the early inflammatory response to β-glucan.
-
Combined Autophagy Inhibition and Dendritic Cell Recruitment Induces Antitumor Immunity and Enhances Immune Checkpoint Blockade Sensitivity in Pancreatic Cancer.
In Cancer Res on 16 December 2024 by Oyama, K., Nakata, K., et al.
PubMed
The effect of immune checkpoint inhibitors is extremely limited in patients with pancreatic ductal adenocarcinoma (PDAC) due to the suppressive tumor immune microenvironment. Autophagy, which has been shown to play a role in antitumor immunity, has been proposed as a therapeutic target for PDAC. In this study, single-cell RNA sequencing of autophagy-deficient murine PDAC tumors revealed that autophagy inhibition in cancer cells induced dendritic cell (DC) activation. Analysis of human PDAC tumors substantiated a negative correlation between autophagy and DC activation signatures. Mechanistically, autophagy inhibition increased the intracellular accumulation of tumor antigens, which could activate DCs. Administration of chloroquine, an autophagy inhibitor, in combination with Flt3 ligand-induced DC infiltration inhibited tumor growth and increased tumor-infiltrating T lymphocytes. However, autophagy inhibition in cancer cells also induced CD8+ T-cell exhaustion with high expression of immune checkpoint LAG3. A triple-therapy comprising chloroquine, Flt3 ligand, and an anti-LAG3 antibody markedly reduced tumor growth in orthotopic syngeneic PDAC mouse models. Thus, targeting autophagy in cancer cells and activating DCs sensitize PDAC tumors to immune checkpoint inhibitor therapy, warranting further development of this treatment approach to overcome immunosuppression in pancreatic cancer. Significance: Inhibiting autophagy in pancreatic cancer cells enhances intracellular accumulation of tumor antigens to induce dendritic cell activation and synergizes with immunotherapy to markedly inhibit the growth of pancreatic ductal adenocarcinoma.
-
Targeting KDM4 family epigenetically triggers antitumour immunity via enhancing tumour-intrinsic innate sensing and immunogenicity.
In Clin Transl Med on 1 February 2024 by Sun, M., Han, X., et al.
PubMed
Despite the remarkable clinical efficacy of cancer immunotherapy, considerable patients fail to benefit from it due to primary or acquired resistance. Tumours frequently hijack diverse epigenetic mechanisms to evade immune detection, thereby highlighting the potential for pharmacologically targeting epigenetic regulators to restore the impaired immunosurveillance and re-sensitise tumours to immunotherapy. Herein, we demonstrated that KDM4-targeting chemotherapeutic drug JIB-04, epigenetically triggered the tumour-intrinsic innate immune responses and immunogenic cell death (ICD), resulting in impressive antitumour effects. Specifically, JIB-04 induced H3K9 hypermethylation through specific inhibition of the KDM4 family (KDM4A-D), leading to impaired DNA repair signalling and subsequent DNA damage. As a result, JIB-04 not only activated the tumour-intrinsic cyclic GMP-AMP synthase (cGAS)-STING pathway via DNA-damage-induced cytosolic DNA accumulation, but also promoted ICD, releasing numerous damage-associated molecular patterns. Furthermore, JIB-04 induced adaptive resistance through the upregulation of programmed death-ligand 1 (PD-L1), which could be overcome with additional PD-L1 blockade. In human tumours, KDM4B expression was negatively correlated with clinical outcomes, type I interferon signatures, and responses to immunotherapy. In conclusion, our results demonstrate that targeting KDM4 family can activate tumour-intrinsic innate sensing and immunogenicity, and synergise with immunotherapy to improve antitumour outcomes.