InVivoMAb anti-mouse PD-1 (CD279)
Product Description
Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Syrian Hamster BKH cells transfected with mouse PD-1 cDNA |
| Reported Applications |
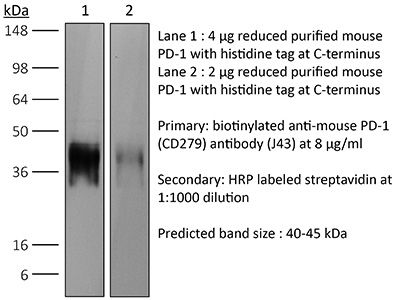
in vivo blocking of PD-1/PD-L signaling in vitro PD-1 neutralization Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107747 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Park, S. J., et al (2014). "Negative role of inducible PD-1 on survival of activated dendritic cells" J Leukoc Biol 95(4): 621-629.
PubMed
PD-1 is a well-established negative regulator of T cell responses by inhibiting proliferation and cytokine production of T cells via interaction with its ligands, B7-H1 (PD-L1) and B7-DC (PD-L2), expressed on non-T cells. Recently, PD-1 was found to be expressed in innate cells, including activated DCs, and plays roles in suppressing production of inflammatory cytokines. In this study, we demonstrate that PD-1 KO DCs exhibited prolonged longevity compared with WT DCs in the dLNs after transfer of DCs into hind footpads. Interestingly, upon LPS stimulation, WT DCs increased the expression of PD-1 and started to undergo apoptosis. DCs, in spleen of LPS-injected PD-1 KO mice, were more resistant to LPS-mediated apoptosis in vivo than WT controls. Moreover, treatment of blocking anti-PD-1 mAb during DC maturation resulted in enhanced DC survival, suggesting that PD-1:PD-L interactions are involved in DC apoptosis. As a result, PD-1-deficient DCs augmented T cell responses in terms of antigen-specific IFN-gamma production and proliferation of CD4 and CD8 T cells to a greater degree than WT DCs. Moreover, PD-1 KO DCs exhibited increased MAPK1 and CD40-CD40L signaling, suggesting a possible mechanism for enhanced DC survival in the absence of PD-1 expression. Taken together, our findings further extend the function of PD-1, which plays an important role in apoptosis of activated DCs and provides important implications for PD-1-mediated immune regulation.
-
Rabenstein, H., et al (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517.
PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
-
Noval Rivas, M., et al (2009). "Reviving function in CD4+ T cells adapted to persistent systemic antigen" J Immunol 183(7): 4284-4291.
PubMed
In bone marrow-transplanted patients, chronic graft-versus-host disease is a complication that results from the persistent stimulation of recipient minor histocompatibility Ag (mHA)-specific T cells contained within the graft. In this study, we developed a mouse model where persistent stimulation of donor T cells by recipient’s mHA led to multiorgan T cell infiltration. Exposure to systemic mHA, however, deeply modified T cell function and chronically stimulated T cells developed a long-lasting state of unresponsiveness, or immune adaptation, characterized by their inability to mediate organ immune damages in vivo. However, analysis of the gene expression profile of adapted CD4+ T cells revealed the specific coexpression of genes known to promote differentiation and function of Th1 effector cells as well as genes coding for proteins that control T cell activity, such as cell surface-negative costimulatory molecules and regulatory cytokines. Strikingly, blockade of negative costimulation abolished T cell adaptation and stimulated strong IFN-gamma production and severe multiorgan wasting disease. Negative costimulation was also shown to control lethal LPS-induced toxic shock in mice with adapted T cells, as well as the capacity of adapted T cells to reject skin graft. Our results demonstrate that negative costimulation is the molecular mechanism used by CD4+ T cells to adapt their activity in response to persistent antigenic stimulation. The effector function of CD4+ T cells that have adapted to chronic Ag presentation can be activated by stimuli strong enough to overcome regulatory signals delivered to the T cells by negative costimulation.
-
Sarraj, B., et al (2014). "Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection" Proc Natl Acad Sci U S A 111(33): 12145-12150.
PubMed
Selectin-selectin ligand interactions mediate the initial steps in leukocyte migration, an integral part of immune responses. Fucosyltransferase-VII (FucT-VII), encoded by Fut7, is essential for biosynthesis of selectin ligands. In an established model of cardiac allograft vasculopathy and chronic rejection, Fut7(-/-) recipients exhibited long-term graft survival with minimal vasculopathy compared with WT controls. Graft survival was associated with CD4 T-cell exhaustion in the periphery, characterized by impaired effector cytokine production, defective proliferation, increased expression of inhibitory receptors programmed death-1 (PD-1) and T cell Ig- and mucin-domain-containing molecule-3 (Tim-3), low levels of IL-7Ralpha on CD4 T cells, and reduced migration of polyfunctional CD4 memory T cells to the allograft. Blocking PD-1 triggered rejection only in Fut7(-/-) recipients, whereas depleting regulatory T cells had no effect in either Fut7(-/-) or WT recipients. Adoptive transfer experiments confirmed that this CD4 T cell-exhausted phenotype is seen primarily in Fut7(-/-) CD4 T cells. These data suggest that impaired leukocyte recruitment is a novel mechanism leading to CD4 T-cell exhaustion. Our experimental system serves as an excellent model to study CD4 T-cell exhaustion as a dominant mechanism of transplant tolerance. Further, targeting FucT-VII may serve as a promising strategy to prevent chronic allograft rejection and promote tolerance.
Product Citations
-
BMP9 potentiates immunotherapy in triple-negative breast cancer by suppressing Tregs infiltration via the PRKDC-CCL2 axis.
In Cancer Lett on 28 February 2026 by You, Y., Wei, L., et al.
PubMed
Immunotherapy represents a pivotal strategy for triple-negative breast cancer (TNBC), yet its efficacy is constrained by the immunosuppressive tumor microenvironment (TME). In this study, we demonstrate that bone morphogenetic protein-9 (BMP9) inhibits tumor growth and reprograms the immune TME in orthotopic TNBC models, primarily by attenuating regulatory T cells (Tregs) infiltration. Tregs depletion abrogates si-BMP9-mediated tumor promotion. Mechanistically, BMP9 suppresses CCL2 expression in an exocrine-independent manner to restrict Tregs recruitment. We identify DNA-dependent protein kinase catalytic subunit (PRKDC) as a BMP9-binding transcriptional regulator. The interaction between PRKDC and BMP9 directly impedes CCL2 transcriptional activation by suppressing PRKDC phosphorylation and indirectly suppresses CCL2 expression via NF-κB pathway remodeling. Critically, BMP9 modulation and CCL2 targeting potentiates immunotherapy efficacy without observable toxicity. Our study unveils the BMP9-PRKDC-CCL2 axis as a master regulatory node for TNBC-Tregs crosstalk, providing a strategy to overcome immunotherapy resistance in TNBC.
-
CD47 blockade (ALX301) enhances immunoradiotherapy response in HPV negative head and neck squamous cell carcinoma.
In PLoS One on 17 February 2026 by Monther, A., Al-Msari, R., et al.
PubMed
Head and neck squamous cell carcinoma (HNSCC) is a significant cause of morbidity and mortality worldwide, with limited treatment options for patients with locally advanced disease. CD47 immune checkpoint inhibitors have been used to block the CD47/SIRPa interaction that inhibits antigen-presenting cell phagocytosis, thereby enhancing antigen presentation to cytotoxic T-cells, and have shown promise in combination with anti-PD1 immunotherapy in tumors, including recurrent/metastatic HNSCC. We found that CD47 expression is associated with poor prognosis in HNSCC and explored the anti-tumor activity of an anti-CD47 fusion protein in combination with anti-PD1 and lymphatic-sparing radiotherapy in a locally advanced HNSCC model. In the 4MOSC1 syngeneic HPV-negative HNSCC mouse model, ALX301 (an engineered CD47-blocking SIRPα fusion for murine models) induced complete tumor regression when combined with anti-PD-1, and produced a partial tumor response as a monotherapy. An anti-PD1 immune checkpoint inhibitor in a CD47-null tumor background led to complete tumor regression confirming a key role for CD47 in tumor immunity. ALX301 treated mice demonstrated increased MHC-II expression on dendritic cells within the tumor and upregulation of CD86 co-stimulatory molecule on dendritic cells within the tumor, sentinel lymph nodes, and contralateral lymph nodes. Combination ALX301 and anti-PD1 treatment in an anti-PD1 resistant 4MOSC2 model demonstrated significant tumor regression, enhanced survivability, improved response with neoadjuvant radiotherapy, and greater retention of CD8 + T-cells within the tumor microenvironment. Notably, T-cell receptor sequencing revealed increased shared clonality between the tumor and sentinel lymph nodes of ALX301 treated mice. These data demonstrate that a combination of CD47 blockade and anti-PD1 therapy enhances tumor antigen presentation and immune cell infiltration, while further improving anti-tumor responses in combination with tumor-targeted radiotherapy. This study provides support for the rational design of combinatorial immunoradiotherapy, using anti-CD47 inhibitors and anti-PD1 therapy, in a clinical trial targeting locally advanced HPV-negative HNSCC.
-
Glycyrrhiza uralensis polysaccharides as a DC-based Vaccine Adjuvant: Enhanced Immunotherapy Combined with PD-1 Blockade
In Research Square on 23 January 2026 by Aili, P., Cai, S., et al.
-
Binary mineral nanoparticles enable intravascular delivery of metal ions to tumors for metalloimmunotherapy.
In Nat Commun on 12 January 2026 by Nguyen, B. L., Le, N. D., et al.
PubMed
Although disruptions in metal ion homeostasis leading to severe cellular damage and regulated cell death are a promising strategy for cancer immunotherapy, challenges in overloading these ions to tumor cells without premature release have limited their therapeutic applications. In this study, we develop binary mineral nanoparticles incorporating both Ca2+ and Na+ ions to enhance the cytotoxic effects of ion interference in cancer immunotherapy. Engineered using a microfluidic system for uniform size distribution and scalability, these nanoparticles exhibit pH-sensitive ion release. Systemically administered, they preferentially accumulate in tumors, elevating intracellular Ca2+ and Na+ levels and inducing immunogenic cell death without calcium channel activators or other small-molecule inducers. Our binary mineral nanoparticles significantly enhance antitumor immunity, especially when combined with an immune checkpoint inhibitor, leading to long-term immunity and inhibition of metastasis. This nanotechnology-enabled synergistic delivery of Ca²⁺ and Na⁺ ions represents a promising adjunct to existing metalloimmunotherapy strategies for cancer eradication.