InVivoMAb anti-mouse Ly6G/Ly6C
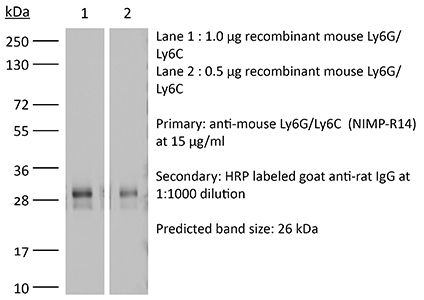
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Purified BALB/c mouse neutrophils |
| Reported Applications |
in vivo neutrophil depletion Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Immunofluorescence Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_2819047 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Castell, S. D., et al (2019). "Neutrophils Which Migrate to Lymph Nodes Modulate CD4(+) T Cell Response by a PD-L1 Dependent Mechanism" Front Immunol 10: 105.
PubMed
It is well known that neutrophils are rapidly recruited to a site of injury or infection and perform a critical role in pathogen clearance and inflammation. However, they are also able to interact with and regulate innate and adaptive immune cells and some stimuli induce the migration of neutrophils to lymph nodes (LNs). Previously, we demonstrated that the immune complex (IC) generated by injecting OVA into the footpad of OVA/CFA immunized mice induced the migration of OVA(+) neutrophils to draining LNs (dLNs). Here we investigate the effects of these neutrophils which reach dLNs on CD4(+) T cell response. Our findings here strongly support a dual role for neutrophils in dLNs regarding CD4(+) T cell response modulation. On the one hand, the CD4(+) T cell population expands after the influx of OVA(+) neutrophils to dLNs. These CD4(+) T cells enlarge their proliferative response, activation markers and IL-17 and IFN-gamma cytokine production. On the other hand, these neutrophils also restrict CD4(+) T cell expansion. The neutrophils in the dLNs upregulate PD-L1 molecules and are capable of suppressing CD4(+) T cell proliferation. These results indicate that neutrophils migration to dLNs have an important role in the homeostasis of adaptive immunity. This report describes for the first time that the influx of neutrophils to dLNs dependent on IC presence improves CD4(+) T cell response, at the same time controlling CD4(+) T cell proliferation through a PD-L1 dependent mechanism.
-
Stackowicz, J., et al (2018). "Evidence that neutrophils do not promote Echis carinatus venom-induced tissue destruction" Nat Commun 9(1): 2304.
PubMed
-
Karmakar, M., et al (2016). "Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP" Nat Commun 7: 10555.
PubMed
Although extracellular ATP is abundant at sites of inflammation, its role in activating inflammasome signalling in neutrophils is not well characterized. In the current study, we demonstrate that human and murine neutrophils express functional cell-surface P2X7R, which leads to ATP-induced loss of intracellular K(+), NLRP3 inflammasome activation and IL-1beta secretion. ATP-induced P2X7R activation caused a sustained increase in intracellular [Ca(2+)], which is indicative of P2X7R channel opening. Although there are multiple polymorphic variants of P2X7R, we found that neutrophils from multiple donors express P2X7R, but with differential efficacies in ATP-induced increase in cytosolic [Ca(2+)]. Neutrophils were also the predominant P2X7R-expressing cells during Streptococcus pneumoniae corneal infection, and P2X7R was required for bacterial clearance. Given the ubiquitous presence of neutrophils and extracellular ATP in multiple inflammatory conditions, ATP-induced P2X7R activation and IL-1beta secretion by neutrophils likely has a significant, wide ranging clinical impact.
-
Karmakar, M., et al (2012). "Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1" J Immunol 189(9): 4231-4235.
PubMed
To examine the role of caspase-1 and the NLRC4 inflammasome during bacterial infection, C57BL/6, IL-1beta(-/-), caspase-1(-/-), and NLRC4(-/-) mouse corneas were infected with ExoS/T- or ExoU-expressing Pseudomonas aeruginosa. We found that IL-1beta was essential for neutrophil recruitment and bacterial clearance and was produced by myeloid cells rather than resident cells. In addition, neutrophils were found to be the primary source of mature IL-1beta during infection, and there was no significant difference in IL-1beta processing between C57BL/6 and caspase-1(-/-) or NLRC4(-/-) infected corneas. IL-1beta cleavage by human and mouse neutrophils was blocked by serine protease inhibitors and was impaired in infected neutrophil elastase (NE)(-/-) corneas. NE(-/-) mice also had an impaired ability to clear the infection. Together, these results demonstrate that during P. aeruginosa infection, neutrophils are the primary source of mature IL-1beta and that IL-1beta processing is dependent on serine proteases and not NLRC4 or caspase-1.
Product Citations
-
The type 2 cytokine Fc-IL-4 revitalizes exhausted CD8+ T cells against cancer.
In Nature on 1 October 2024 by Feng, B., Bai, Z., et al.
PubMed
Current cancer immunotherapy predominately focuses on eliciting type 1 immune responses fighting cancer; however, long-term complete remission remains uncommon1,2. A pivotal question arises as to whether type 2 immunity can be orchestrated alongside type 1-centric immunotherapy to achieve enduring response against cancer3,4. Here we show that an interleukin-4 fusion protein (Fc-IL-4), a typical type 2 cytokine, directly acts on CD8+ T cells and enriches functional terminally exhausted CD8+ T (CD8+ TTE) cells in the tumour. Consequently, Fc-IL-4 enhances antitumour efficacy of type 1 immunity-centric adoptive T cell transfer or immune checkpoint blockade therapies and induces durable remission across several syngeneic and xenograft tumour models. Mechanistically, we discovered that Fc-IL-4 signals through both signal transducer and activator of transcription 6 (STAT6) and mammalian target of rapamycin (mTOR) pathways, augmenting the glycolytic metabolism and the nicotinamide adenine dinucleotide (NAD) concentration of CD8+ TTE cells in a lactate dehydrogenase A-dependent manner. The metabolic modulation mediated by Fc-IL-4 is indispensable for reinvigorating intratumoural CD8+ TTE cells. These findings underscore Fc-IL-4 as a potent type 2 cytokine-based immunotherapy that synergizes effectively with type 1 immunity to elicit long-lasting responses against cancer. Our study not only sheds light on the synergy between these two types of immune responses, but also unveils an innovative strategy for advancing next-generation cancer immunotherapy by integrating type 2 immune factors.
-
West Nile virus triggers intestinal dysmotility via T cell-mediated enteric nervous system injury.
In J Clin Invest on 29 August 2024 by Janova, H., Zhao, F. R., et al.
PubMed
Intestinal dysmotility syndromes have been epidemiologically associated with several antecedent bacterial and viral infections. To model this phenotype, we previously infected mice with the neurotropic flavivirus West Nile virus (WNV) and demonstrated intestinal transit defects. Here, we found that within 1 week of WNV infection, enteric neurons and glia became damaged, resulting in sustained reductions of neuronal cells and their networks of connecting fibers. Using cell-depleting antibodies, adoptive transfer experiments, and mice lacking specific immune cells or immune functions, we show that infiltrating WNV-specific CD4+ and CD8+ T cells damaged the enteric nervous system (ENS) and glia, which led to intestinal dysmotility; these T cells used multiple and redundant effector molecules including perforin and Fas ligand. In comparison, WNV-triggered ENS injury and intestinal dysmotility appeared to not require infiltrating monocytes, and damage may have been limited by resident muscularis macrophages. Overall, our experiments support a model in which antigen-specific T cell subsets and their effector molecules responding to WNV infection direct immune pathology against enteric neurons and supporting glia that results in intestinal dysmotility.
-
Tumour-derived Extracellular Vesicle and Particle Reprogramming of Interstitial Macrophages in the Lung Pre-Metastatic Niche Enhances Vascular Permeability and Metastatic Potential
In Research Square on 30 May 2024 by Lyden, D., Dror, S., et al.
-
Heterodimerization of T cell engaging bispecific antibodies to enhance specificity against pancreatic ductal adenocarcinoma.
In J Hematol Oncol on 23 April 2024 by Long, A. W., Xu, H., et al.
PubMed
EGFR and/or HER2 expression in pancreatic cancers is correlated with poor prognoses. We generated homodimeric (EGFRxEGFR or HER2xHER2) and heterodimeric (EGFRxHER2) T cell-engaging bispecific antibodies (T-BsAbs) to direct polyclonal T cells to these antigens on pancreatic tumors.