InVivoMAb anti-mouse CD4
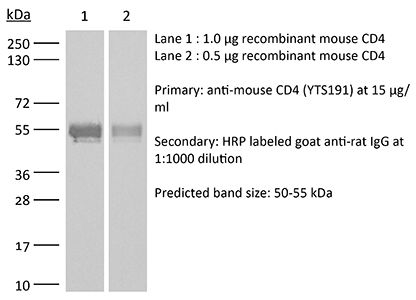
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications | in vivo CD4+ T cell depletion |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950382 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Burrack, K. S., et al (2015). "Myeloid Cell Arg1 Inhibits Control of Arthritogenic Alphavirus Infection by Suppressing Antiviral T Cells" PLoS Pathog 11(10): e1005191.
PubMed
Arthritogenic alphaviruses, including Ross River virus (RRV) and chikungunya virus (CHIKV), are responsible for explosive epidemics involving millions of cases. These mosquito-transmitted viruses cause inflammation and injury in skeletal muscle and joint tissues that results in debilitating pain. We previously showed that arginase 1 (Arg1) was highly expressed in myeloid cells in the infected and inflamed musculoskeletal tissues of RRV- and CHIKV-infected mice, and specific deletion of Arg1 from myeloid cells resulted in enhanced viral control. Here, we show that Arg1, along with other genes associated with suppressive myeloid cells, is induced in PBMCs isolated from CHIKV-infected patients during the acute phase as well as the chronic phase, and that high Arg1 expression levels were associated with high viral loads and disease severity. Depletion of both CD4 and CD8 T cells from RRV-infected Arg1-deficient mice restored viral loads to levels detected in T cell-depleted wild-type mice. Moreover, Arg1-expressing myeloid cells inhibited virus-specific T cells in the inflamed and infected musculoskeletal tissues, but not lymphoid tissues, following RRV infection in mice, including suppression of interferon-gamma and CD69 expression. Collectively, these data enhance our understanding of the immune response following arthritogenic alphavirus infection and suggest that immunosuppressive myeloid cells may contribute to the duration or severity of these debilitating infections.
-
Wensveen, F. M., et al (2015). "NK cells link obesity-induced adipose stress to inflammation and insulin resistance" Nat Immunol 16(4): 376-385.
PubMed
An important cause of obesity-induced insulin resistance is chronic systemic inflammation originating in visceral adipose tissue (VAT). VAT inflammation is associated with the accumulation of proinflammatory macrophages in adipose tissue, but the immunological signals that trigger their accumulation remain unknown. We found that a phenotypically distinct population of tissue-resident natural killer (NK) cells represented a crucial link between obesity-induced adipose stress and VAT inflammation. Obesity drove the upregulation of ligands of the NK cell-activating receptor NCR1 on adipocytes; this stimulated NK cell proliferation and interferon-gamma (IFN-gamma) production, which in turn triggered the differentiation of proinflammatory macrophages and promoted insulin resistance. Deficiency of NK cells, NCR1 or IFN-gamma prevented the accumulation of proinflammatory macrophages in VAT and greatly ameliorated insulin sensitivity. Thus NK cells are key regulators of macrophage polarization and insulin resistance in response to obesity-induced adipocyte stress.
-
Kish, D. D., et al (2009). "CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge" J Immunol 182(10): 5949-5959.
PubMed
Effector CD8 T cell recruitment into the skin in response to Ag challenge requires prior CXCL1/KC-directed neutrophil infiltration. Mechanisms inducing CXCL1 production and the dynamics of neutrophil-CD8 T cell interactions during elicitation of Ag-specific responses in the skin were investigated. CXCL1 and CXCL2/MIP-2 were produced within 3-6 h of Ag challenge at 10-fold higher levels in skin challenge sites of Ag-sensitized vs nonsensitized mice. In the challenge sites of sensitized mice this production decreased at 6-9 h postchallenge to near the levels observed in skin challenge sites of nonsensitized mice but rose to a second peak 12 h after challenge. The elevated early neutrophil chemoattractant production at 3-6 h after skin challenge of sensitized animals required both IFN-gamma and IL-17, produced by distinct populations of Ag-primed CD8 T cells in response to Ag challenge. Although induced by the Ag-primed CD8 T cells, the early CXCL1 and CXCL2 production was accompanied by neutrophil but not CD8 T cell infiltration into the skin Ag challenge site. Infiltration of the CD8 T cells into the challenge site was not observed until 18-24 h after challenge. These results demonstrate an intricate series of early interactions between Ag-specific and innate immune components that regulate the sequential infiltration of neutrophils and then effector T cells into the skin to mediate an immune response.
-
Yamada, D. H., et al (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390.
PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
Product Citations
-
A novel mRNA-based therapeutic vaccine elicits robust anti-tumor immunity against HPV-associated malignancies.
In Front Immunol on 4 May 2026 by Li, Q., Liu, Y., et al.
PubMed
Human papillomavirus (HPV) infection is strongly associated with multiple malignancies, primarily driven by the viral oncoproteins E6 and E7, which play a central role in HPV-induced malignant transformation. Although current prophylactic HPV vaccines have shown remarkable efficacy in preventing initial infections, there remains an urgent need for therapeutic vaccines targeting pre-existing HPV infections and HPV-associated malignancies.
-
The integrated stress response promotes immune evasion through lipocalin 2.
In Nature on 1 April 2026 by Bossowski, J. P., Pillai, R., et al.
PubMed
Cancer cells activate the integrated stress response (ISR) to adapt to stress and resist therapy1. ISR signals converge on activating transcription factor 4 (ATF4), which controls cell-intrinsic transcriptional programs that are involved in metabolic adaptation, survival and growth2,3. However, whether the ISR-ATF4 axis influences anti-tumour immune responses remains mostly unknown. Here we show that loss of ATF4 decreases tumour progression considerably in immunocompetent mice, but not in immunocompromised ones, by enhancing T cell-dependent anti-cancer immune responses. An unbiased genetic screen of ATF4-regulated genes identifies lipocalin 2 (LCN2) as the principal ATF4-dependent effector that impairs anti-tumour immunity by favouring infiltration with immunosuppressive interstitial macrophages. Furthermore, we find that LCN2 promotes T cell exclusion and immune evasion in preclinical mouse models, and correlates with decreased T cell infiltration in patients with lung and pancreatic adenocarcinomas. Anti-LCN2 antibodies promote robust anti-tumour T cell responses in mouse models of aggressive solid tumours. Our study shows that the ATF4-LCN2 axis has a cell-extrinsic role in suppressing anti-cancer immunity, and could pave the way for an immunotherapy approach that targets LCN2.
-
Uridine depletion impairs CD8⁺ T cell antitumor activity through N-glycosylation.
In Cell Metab on 3 March 2026 by Xiao, J., Li, Z., et al.
PubMed
Immune checkpoint blockade (ICB) faces limitations owing to high cost and restricted efficacy. This study identifies SNX17 as a key mediator of ICB resistance. Elevated SNX17 correlates with poor anti-PD-1 response in humans and mice. SNX17 deletion in tumor cells inhibits tumor growth via CD8+ T cell-dependent mechanisms. SNX17 reduces uridine in the tumor microenvironment (TME), suppressing IFN-γ and upregulating PD1 in CD8+ T cells. Exogenous uridine shows antitumor efficacy comparable to anti-PD-1/PD-L1 in low-SNX17 tumors and overcomes resistance in high-SNX17 models. Uridine enhances CD8+ T cell function by promoting CD45 N-glycosylation and LCK phosphorylation. Mechanistically, SNX17 stabilizes RUNX2, promoting UPP1 transcription and uridine degradation in the TME. These findings position SNX17 as an ICB response biomarker and nominate uridine as a cost-effective immunotherapeutic strategy.
-
Disruption of tRNA threonylation triggers RIG-I mediated anti-tumour immune response.
In Nat Commun on 25 February 2026 by Dziagwa, C., Seca, C., et al.
PubMed
Tumour-induced mechanisms of immune evasion hinder immune response to cancer, particularly in melanoma. mRNA translation, by ensuring accurate protein synthesis, regulates cancer phenotypes and immune response, but the underlying mechanisms remain unclear. Here, we reveal how O-sialoglycoprotein endopeptidase (OSGEP), catalysing the tRNA modification N6-threonylcarbamoyladenosine (t6A), drives protein homeostasis in cancer cells to maintain T-cell exclusion and prevent anti-tumour immune response. t6A-deficient melanoma cells disrupt efficient cytoplasmic translation of ANN codons (trinucleotides with A in the first position and N = any nucleotide), causing specific protein aggregation and the formation of integrated stress response-dependent stress granules. We discovered that OSGEP loss triggers melanoma regression by relocating RIG-I to stress granules, leading to its pathway activation. As a result, T-cells are recruited to the tumour site and orchestrate an anti-tumour immune response. Finally, an OSGEP-driven gene signature in melanoma patients is associated with T-cell infiltration and improved overall survival. Together, our findings position t6A tRNA modification as a promising therapeutic target for melanoma treatment.