InVivoMAb anti-mouse CD4
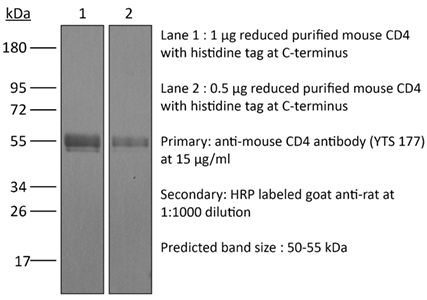
Product Description
Specifications
| Isotype | Rat IgG2a |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse Spleen Cells |
| Reported Applications |
in vivo blockade of CD4+ T-cell responses Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107642 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Li, Z., et al (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129.
PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
-
Mayer, C. T., et al (2014). "Anti-CD4 treatment inhibits autoimmunity in scurfy mice through the attenuation of co-stimulatory signals" J Autoimmun 50: 23-32.
PubMed
A major concept in autoimmunity is that disruption of Foxp3(+) regulatory T cells (Tregs) predisposes to breach of tolerance. This is exemplified by the Foxp3-linked disorder termed IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) which affects newborn children. There has been considerable clinical interest in the role of non-depleting anti-CD4 antibodies as a means of upregulating the function of Foxp3(+) Tregs in order to control detrimental inflammatory responses such as transplant rejection. However, according to the paradigm of a Treg-dependent mechanism of action, the effectiveness of anti-CD4 antibodies as a therapy for human autoimmune diseases is unclear considering that Treg function might be intrinsically impaired. Specifically, anti-CD4 therapy is expected to fail in patients suffering from the IPEX syndrome due to the lack of functional Foxp3(+) Tregs. Taking advantage of natural Foxp3 mutant scurfy (sf) mice closely resembling the IPEX syndrome, and genetically engineered mice depleted of Foxp3(+) Tregs, we report here that anti-CD4 treatment induces tolerance independent of Foxp3(+) Tregs. This so far undefined mechanism is dependent on the recessive non-infectious tolerization of autoreactive T cells. Treg-independent tolerance alone is powerful enough to suppress both the onset and severity of autoimmunity and reduces clinically relevant autoantibody levels and liver fibrosis. Mechanistically, tolerance induction requires the concomitant activation of autoreactive T cells and is associated with the down-regulation of the co-stimulatory TNF-receptor superfamily members OX40 and CD30 sustaining CD4(+) T cell survival. In the light of ongoing clinical trials, our results highlight an unexpected potency of anti-CD4 antibodies for the treatment of autoimmune diseases. Particularly, CD4 blockade might represent a novel therapeutic option for the human IPEX syndrome.
-
Rocca, C. J., et al (2014). "rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: conclusion of a comparative study" Gene Ther 21(6): 618-628.
PubMed
Effective gene therapy strategies for the treatment of kidney disorders remain elusive. We report an optimized kidney-targeted gene delivery strategy using recombinant adeno-associated virus (rAAV) administered via retrograde renal vein injection in mice. Renal vein injection of rAAV consistently resulted in superior kidney transduction compared with tail vein injection using as little as half the tail vein dose. We compared rAAV5, 6, 8 and 9, containing either green fluorescent protein (GFP) or luciferase reporter genes driven by the Cytomegalovirus promoter. We demonstrated that although rAAV6 and 8 injected via renal vein transduced the kidney, transgene expression was mainly restricted to the medulla. Transgene expression was systematically low after rAAV5 injection, attributed to T-cell immune response, which could be overcome by transient immunosuppression. However, rAAV9 was the only serotype that permitted high-transduction efficiency of both the cortex and medulla. Moreover, both the glomeruli and tubules were targeted, with a higher efficiency within the glomeruli. To improve the specificity of kidney-targeted gene delivery with rAAV9, we used the parathyroid hormone receptor ‘kidney-specific’ promoter. We obtained a more efficient transgene expression within the kidney, and a significant reduction in other tissues. Our work represents the first comprehensive and clinically relevant study for kidney gene delivery.
Product Citations
-
PD-1 inhibitor improves radiosensitivity by tumor vessel normalization.
In Br J Cancer on 1 March 2026 by Hao, S., Ai, D., et al.
PubMed
Host immunity status and hypoxia are the hallmarks of radiosensitivity. Induction of anti-PD-1 immunotherapy demonstrates promise in locally advanced tumor radiotherapy, but whether anti-PD-1 immunotherapy improves radiosensitivity is unclear.
-
CBD promotes antitumor activity by modulating tumor immune microenvironment in HPV associated head and neck squamous cell carcinoma.
In Front Immunol on 6 June 2025 by Sen, P., Sadat, S., et al.
PubMed
Marijuana use is associated with HPV-positive head and neck squamous cell carcinoma (HNSCC). However, cannabinoid use continues to increase in the US general population for recreational purposes as well as in cancer patients for palliative care. In this study, we explored the role of cannabidiol (CBD) in promoting anti-tumor activity by modulating immune response in HPV-positive HNSCC by using pre-clinical models.
-
Eph Receptors Activate Myeloid Checkpoint Receptor LILRB5 to Support Tumor Development.
In Cancer Immunol Res on 4 June 2025 by He, Y., Zhang, C., et al.
PubMed
Immunosuppressive myeloid cells are critical obstacles to T cell-centered immune checkpoint blockade therapies, which have been successful in treating a fraction of patients with cancer. How tumor cells interact with myeloid cells to regulate immune responses and tumor development is unclear. In this study, we report that certain membrane tyrosine kinase Eph receptors, including EphA7 and EphB1, specifically bind the immune inhibitory receptors leukocyte Ig-like receptor family B 5 (LILRB5) and LILRB2. These Eph receptors induce LILRB5-mediated signaling activation, and LILRB5 also activates Eph receptor signaling. Activation of LILRB5 promoted immunosuppressive marker expression and inhibited activating marker expression on myeloid cells from patients with cancer in vitro. Upon myeloid cell-specific expression of LILRB5 in transgenic mice, the interaction between the Eph receptor on tumor cells and LILRB5 on myeloid cells led to increased tumor growth, increased immunosuppressive myeloid cells, and decreased frequencies of functional T cells compared with control mice. Eph-induced LILRB5 signaling and functions were reversed by LILRB5 blockade. In sum, certain Eph receptors functionally interact with the myeloid checkpoint receptor LILRB5 resulting in bidirectional signaling, and LILRB5 plays an important role in supporting immunosuppressive myeloid cells and sustaining tumor development.
-
Probiotics and their metabolite spermidine enhance IFN-γ+CD4+ T cell immunity to inhibit hepatitis B virus.
In Cell Rep Med on 19 November 2024 by Wang, T., Fan, Y., et al.
PubMed
The therapeutic potential of commensal microbes and their metabolites is promising in the functional cure of chronic hepatitis B virus (HBV) infection, which is defined as hepatitis B surface antigen (HBsAg) loss. Here, using both specific-pathogen-free and germ-free mice, we report that probiotics significantly promote the decline of HBsAg and inhibit HBV replication by enhancing intestinal homeostasis and provoking intrahepatic interferon (IFN)-γ+CD4+ T cell immune response. Depletion of CD4+ T cells or blockage of IFN-γ abolishes probiotics-mediated HBV inhibition. Specifically, probiotics-derived spermidine accumulates in the gut and transports to the liver, where it exhibits a similar anti-HBV effect. Mechanistically, spermidine enhances IFN-γ+CD4+ T cell immunity by autophagy. Strikingly, administration of probiotics in HBV patients reveals a preliminary trend to accelerate the decline of serum HBsAg. In conclusion, probiotics and their derived spermidine promote HBV clearance via autophagy-enhanced IFN-γ+CD4+ T cell immunity, highlighting the therapeutic potential of probiotics and spermidine for the functional cure of HBV patients.