InVivoMAb anti-mouse CD3ε
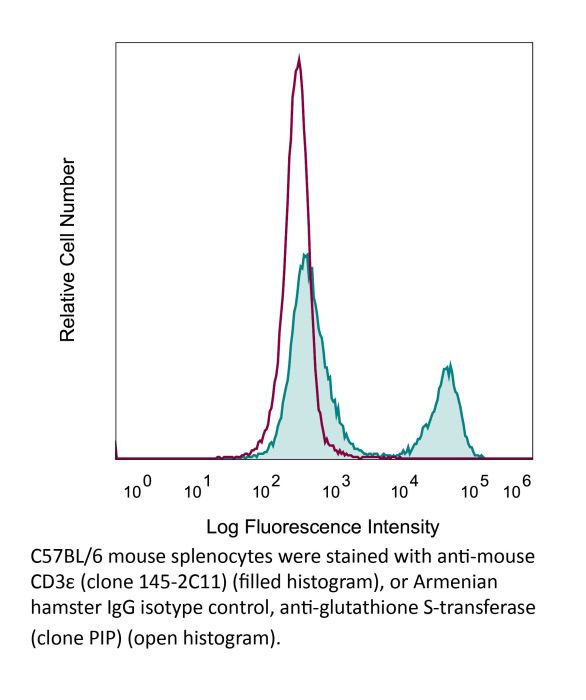
Product Description
Specifications
| Isotype | Armenian Hamster IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse BM10-37 cytotoxic T cells |
| Reported Applications |
in vivo T cell depletion in vitro T cell stimulation/activation Immunofluorescence Flow cytometry Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_1107634 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Tang, W., et al (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566.
PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
-
Berger, H., et al (2013). "SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth" Cancer Res 73(12): 3578-3590.
PubMed
Activation of the transcription factor PPARgamma by the n-3 fatty acid docosahexaenoic acid (DHA) is implicated in controlling proinflammatory cytokine secretion, but the intracellular signaling pathways engaged by PPARgamma are incompletely characterized. Here, we identify the adapter-encoding gene SOCS3 as a critical transcriptional target of PPARgamma. SOCS3 promoter binding and gene transactivation by PPARgamma was associated with a repression in differentiation of proinflammatory T-helper (TH)17 cells. Accordingly, TH17 cells induced in vitro displayed increased SOCS3 expression and diminished capacity to produce interleukin (IL)-17 following activation of PPARgamma by DHA. Furthermore, naive CD4 T cells derived from mice fed a DHA-enriched diet displayed less capability to differentiate into TH17 cells. In two different mouse models of cancer, DHA prevented tumor outgrowth and angiogenesis in an IL-17-dependent manner. Altogether, our results uncover a novel molecular pathway by which PPARgamma-induced SOCS3 expression prevents IL-17-mediated cancer growth.
-
Rabenstein, H., et al (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517.
PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
-
Gu, A. D., et al (2015). "A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling" Immunity 42(1): 68-79.
PubMed
Transforming growth factor-beta (TGF-beta) suppresses T cell function to maintain self-tolerance and to promote tumor immune evasion. Yet how Smad4, a transcription factor component of TGF-beta signaling, regulates T cell function remains unclear. Here we have demonstrated an essential role for Smad4 in promoting T cell function during autoimmunity and anti-tumor immunity. Smad4 deletion rescued the lethal autoimmunity resulting from transforming growth factor-beta receptor (TGF-betaR) deletion and compromised T-cell-mediated tumor rejection. Although Smad4 was dispensable for T cell generation, homeostasis, and effector function, it was essential for T cell proliferation after activation in vitro and in vivo. The transcription factor Myc was identified to mediate Smad4-controlled T cell proliferation. This study thus reveals a requirement of Smad4 for T-cell-mediated autoimmunity and tumor rejection, which is beyond the current paradigm. It highlights a TGF-betaR-independent role for Smad4 in promoting T cell function, autoimmunity, and anti-tumor immunity.
Product Citations
-
IL-6 Exacerbates Experimental Autoimmune Prostatitis by Disrupting STAT5a-Mediated Treg Cell Function and Th17/Treg Balance.
In Mediators Inflamm on 26 May 2026 by Liu, X., Bian, X., et al.
PubMed
Chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS) is a common urological condition in men. Although disruption of the Th17 (T helper 17)/Treg (regulatory T) balance has been implicated in its pathogenesis, the upstream drivers of this immune imbalance remain incompletely understood. This study investigated whether interleukin-6 (IL-6) is associated with altered Th17/Treg homeostasis and impaired Treg function in experimental autoimmune prostatitis (EAP).
-
Preclinical efficacy of dendritic cells loaded with newly identified HPV11 E6-derived CD8+ T cell epitopes.
In NPJ Vaccines on 27 April 2026 by Baert, L., Gu, C., et al.
PubMed
Although the low-risk human papillomaviruses (HPVs), including HPV11, are mainly associated with low-grade lesions, they are also implicated in otolaryngologic malignancies and lung carcinomas. Nonetheless, T cell responses to the low-risk HPVs remain poorly understood. This study investigated HPV11 E6/7-specific T cell responses in C57BL/6 mice challenged with TC-1 tumor cells expressing HPV11 E6/7 proteins (TC-1.HPV11 E6/7). We identified two major CD8+ T cell epitopes, HPV11 E641-55 (AEIYAYAYKNLKVVW, p11) and E685-99 (YAPTVEEETNEDILK, p22). We next demonstrated that IFNα-matured bone marrow-derived dendritic cells (BMDCs) loaded with peptides p11 or p22 induced p11- and p22-specific CD8+ T cell responses, respectively. Furthermore, the administration of p11- or p22-loaded BMDCs matured with IFNα or BMDCs generated in the presence of p38 inhibitor/IL-15 and matured with TNFα, IL-1β, and prostaglandin E2, suppressed TC-1.HPV11.E6/7 tumor progression with increased infiltrations of p11- and p22-specific IFNγ+, TNFα+, and granzyme B+ CD8+ T cells in the tumors. In addition, BMDCs loaded with both peptide epitopes, p11 and p22, were more effective than BMDCs loaded with single epitopes in the suppression of TC-1.HPV11 E6/7 tumor progression. Data from this study will help with the rational design of therapeutic strategies for the diseases associated with HPV11 infection.
-
A senescent tumor cell-derived nanovesicle directly primes splenic T cells to potentiate cancer radiotherapy.
In Cell Rep Med on 21 April 2026 by Pan, J., Zhang, S., et al.
PubMed
Radiotherapy (RT)-induced senescent tumor cells (STCs) reinforce an immunosuppressive tumor microenvironment (ITM) and compromise therapeutic outcomes. However, current senolytic strategies lack specificity for STCs and often cause off-target toxicity. Here, we observe that STCs possess enhanced antigen-presenting capacity in patient-derived tumor tissues and murine tumor models. Leveraging this phenomenon, we engineer STC-derived nanovesicles (termed nano-APM) for preserving endogenous antigens and antigen-presenting cues. We demonstrate that systemically administered nano-APMs accumulate in the spleen and establish a pool of STC-specific CD8+ T cells. Sequential integration of RT induces local tumor senescence, and nano-APMs then effectively mobilize the STC-specific T cells to stimulate a confined recall response. In murine tumor models, the combination of nano-APM plus RT selectively eliminates STCs, reprograms RT-induced ITM, and elicits durable antitumor immunity. Collectively, this study establishes STC-derived nanovesicles as a practical means to enhance RT efficacy by enabling splenic T cell priming and spatiotemporally confined senolysis.
-
Mitochondrial metabolism and signaling direct dendritic cell function in antitumor immunity.
In Science on 2 April 2026 by You, Z., Kim, J., et al.
PubMed
Antitumor immunity requires conventional type 1 dendritic cells (cDC1s). How cDC1s maintain functional fitness in the tumor microenvironment remains unclear. In this study, we established that intratumoral cDC1s exhibited discrete mitochondrial states and that OPA1-mediated mitochondrial energy and redox metabolism dictated cDC1 antitumor responses. Mechanistically, OPA1 orchestrated antigen presentation and the CD8+ T cell priming function of cDC1s by promoting nuclear respiratory factor 1 (NRF1) expression and electron transport chain integrity, thereby supporting bioenergetics and NAD+/NADH balance. During tumor progression, mitochondrial membrane potential and volume, as well as OPA1-NRF1 signaling, declined in intratumoral cDC1s. Furthermore, intratumoral administration of cDC1s with polarized mitochondria showed immunotherapeutic benefits in mice, particularly in combination with immune checkpoint blockade. Collectively, our findings reveal mitochondrial metabolism and signaling as putative targets to reinvigorate cDC1 function for cancer immunotherapy.