InVivoPlus anti-mouse CD3ε
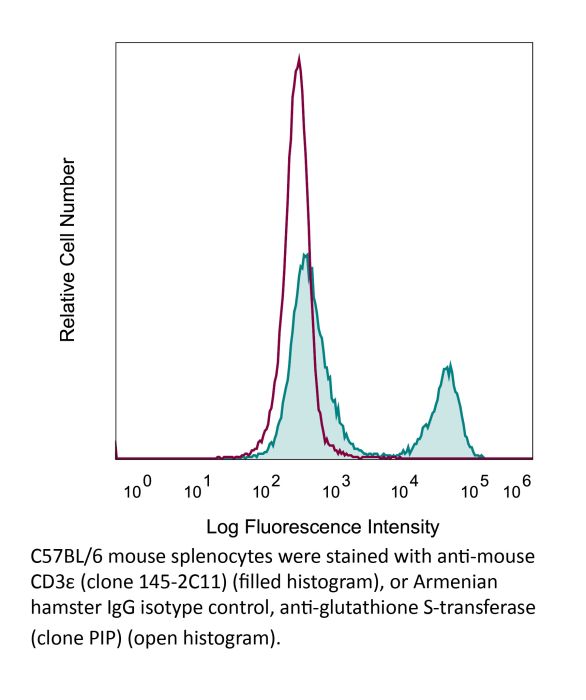
Product Description
Specifications
| Isotype | Armenian Hamster IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse BM10-37 cytotoxic T cells |
| Reported Applications |
in vivo T cell depletion in vitro T cell stimulation/activation Immunofluorescence Flow cytometry Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_1107634 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Tang, W., et al (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566.
PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
-
Berger, H., et al (2013). "SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth" Cancer Res 73(12): 3578-3590.
PubMed
Activation of the transcription factor PPARgamma by the n-3 fatty acid docosahexaenoic acid (DHA) is implicated in controlling proinflammatory cytokine secretion, but the intracellular signaling pathways engaged by PPARgamma are incompletely characterized. Here, we identify the adapter-encoding gene SOCS3 as a critical transcriptional target of PPARgamma. SOCS3 promoter binding and gene transactivation by PPARgamma was associated with a repression in differentiation of proinflammatory T-helper (TH)17 cells. Accordingly, TH17 cells induced in vitro displayed increased SOCS3 expression and diminished capacity to produce interleukin (IL)-17 following activation of PPARgamma by DHA. Furthermore, naive CD4 T cells derived from mice fed a DHA-enriched diet displayed less capability to differentiate into TH17 cells. In two different mouse models of cancer, DHA prevented tumor outgrowth and angiogenesis in an IL-17-dependent manner. Altogether, our results uncover a novel molecular pathway by which PPARgamma-induced SOCS3 expression prevents IL-17-mediated cancer growth.
-
Rabenstein, H., et al (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517.
PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
-
Gu, A. D., et al (2015). "A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling" Immunity 42(1): 68-79.
PubMed
Transforming growth factor-beta (TGF-beta) suppresses T cell function to maintain self-tolerance and to promote tumor immune evasion. Yet how Smad4, a transcription factor component of TGF-beta signaling, regulates T cell function remains unclear. Here we have demonstrated an essential role for Smad4 in promoting T cell function during autoimmunity and anti-tumor immunity. Smad4 deletion rescued the lethal autoimmunity resulting from transforming growth factor-beta receptor (TGF-betaR) deletion and compromised T-cell-mediated tumor rejection. Although Smad4 was dispensable for T cell generation, homeostasis, and effector function, it was essential for T cell proliferation after activation in vitro and in vivo. The transcription factor Myc was identified to mediate Smad4-controlled T cell proliferation. This study thus reveals a requirement of Smad4 for T-cell-mediated autoimmunity and tumor rejection, which is beyond the current paradigm. It highlights a TGF-betaR-independent role for Smad4 in promoting T cell function, autoimmunity, and anti-tumor immunity.
Product Citations
-
Metabolically reprogrammed eosinophils impair T cell immunity and cause chronic skin infection.
In EMBO Mol Med on 1 April 2026 by Barinberg, D., Sebald, H., et al.
PubMed
Eosinophils exhibit antimicrobial, cytotoxic and immunoregulatory effects, but our knowledge of their transcriptional and functional heterogeneity is still limited, especially in non-intestinal tissues. Here, we used a mouse model of chronic cutaneous inflammation elicited by the protozoan pathogen Leishmania mexicana to investigate the function and transcriptional dynamics of skin eosinophils. Infection of C57BL/6 mice triggered local and systemic eosinophilia that was driven by type 2 innate lymphoid cells and interleukin-5. Genetic and pharmacological eosinophil depletion led to an enhanced Th1 response, polarization towards M1-like macrophages and resolution of clinical disease, despite an unexpected simultaneous upregulation of IL-4. Single-cell transcriptomics revealed a skin-imprinted trajectory of inflammatory eosinophils that strongly expressed the glucose transporter Slc2a3 (GLUT3) These eosinophils impeded the function of Th1 cells by forming a competitive metabolic niche through preferential glucose uptake. Our findings uncover an inflammatory, metabolically reprogrammed eosinophil population that promotes chronic skin inflammation by limiting protective T cell responses.
-
A conserved eIF1A+ luminal cell-centered hypoxic and "cold" tumor microenvironment promotes pan-subtype prostate cancer progression.
In Cell Rep Med on 17 February 2026 by Cheng, Y., Wan, L., et al.
PubMed
Prostate cancer (PCa) is a malignancy with high heterogeneity arising from tumor microenvironment and histological subtypes. Identifying conserved progression drivers within such heterogeneity is essential for improving clinical outcomes. Using imaging mass cytometry, this study analyzes 38 proteins across paracancerous tissue and four histological subtypes: low-grade prostate acinar adenocarcinoma (LgPAC), high-grade PAC (HgPAC), intraductal carcinoma (IDC), and ductal adenocarcinoma (DAC). Results reveal that eIF1A is overexpressed in high-risk subtypes including HgPAC, IDC, and DAC and correlates with poor prognosis. In luminal cells, EIF1A knockdown and the translation inhibitor homoharringtonine (HHT) both suppress HIF-1α translation and tumor growth, while promoting infiltration of anticancer immune cells including PD-1- T cells and CD163- macrophages. Clinically, neoadjuvant HHT combined with androgen deprivation therapy reduces hypoxia and enhances immune cell infiltration, as shown by single-cell RNA sequencing. Collectively, this work defines conserved molecular features across PCa subtypes, providing promising insights for clinical management. This study was registered at Clinicaltrials.gov (NCT06834321).
-
TMEM41B is an endoplasmic reticulum Ca2+ release channel maintaining naive T cell quiescence and responsiveness.
In Cell Discov on 4 March 2025 by Ma, Y., Wang, Y., et al.
PubMed
In mammalian cells, endoplasmic reticulum (ER) passively releases Ca2+ under steady state, but channels involved remain elusive. Here, we report that TMEM41B, an ER-resident membrane protein critical for autophagy, lipid metabolism, and viral infection, functions as an ER Ca2+ release channel. Biochemically, purified recombinant TMEM41B forms a concentration-dependent Ca2+ channel in single-channel electrophysiology assays. Cellularly, TMEM41B deficiency causes ER Ca2+ overload, while overexpression of TMEM41B depletes ER Ca2+. Immunologically, ER Ca2+ overload leads to upregulation of IL-2 and IL-7 receptors in naive T cells, which in turn increases basal signaling of JAK-STAT, AKT-mTOR, and MAPK pathways. This dysregulation drives TMEM41B-deficient naive T cells into a metabolically activated yet immunologically naive state. ER Ca2+ overload also downregulates CD5, lowering the activation threshold of TMEM41B-deficient T cells and leading to heightened T cell responses during infections. In summary, we identify TMEM41B as a concentration-dependent ER Ca2+ release channel, revealing an unexpected role of ER Ca2+ in naive T cell quiescence and responsiveness.
-
Short-chain fatty acids are a key mediator of gut microbial regulation of T cell trafficking and differentiation after traumatic brain injury
In Research Square on 21 November 2024 by Celorrio, M., Shumilov, K., et al.