InVivoMAb anti-human/monkey CD28
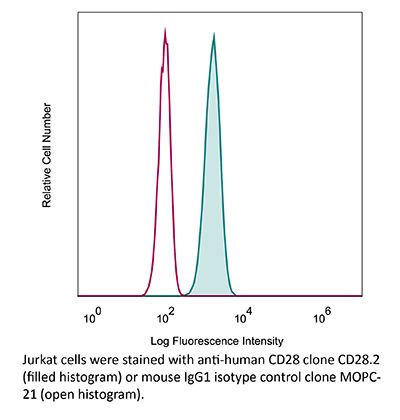
Product Description
Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Human CD28 transfected cell line |
| Reported Applications |
in vitro T cell stimulation/activation Immunoprecipitation Flow cytometry Immunohistochemistry (frozen) |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687814 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Zhao, Y., et al (2019). "PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways" Immunity 51(6): 1059-1073.e1059.
PubMed
Combined immunotherapy targeting the immune checkpoint receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1), or CTLA-4 and the PD-1 ligand (PD-L1) exhibits superior anti-tumor responses compared with single-agent therapy. Here, we examined the molecular basis for this synergy. Using reconstitution assays with fluorescence readouts, we found that PD-L1 and the CTLA-4 ligand CD80 heterodimerize in cis but not trans. Quantitative biochemistry and cell biology assays revealed that PD-L1:CD80 cis-heterodimerization inhibited both PD-L1:PD-1 and CD80:CTLA-4 interactions through distinct mechanisms but preserved the ability of CD80 to activate the T cell co-stimulatory receptor CD28. Furthermore, PD-L1 expression on antigen-presenting cells (APCs) prevented CTLA-4-mediated trans-endocytosis of CD80. Atezolizumab (anti-PD-L1), but not anti-PD-1, reduced cell surface expression of CD80 on APCs, and this effect was negated by co-blockade of CTLA-4 with ipilimumab (anti-CTLA-4). Thus, PD-L1 exerts an immunostimulatory effect by repressing the CTLA-4 axis; this has implications to the synergy of anti-PD-L1 and anti-CTLA-4 combination therapy.
-
Blewett, M. M., et al (2016). "Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells" Sci Signal 9(445): rs10.
PubMed
Dimethyl fumarate (DMF) is an electrophilic drug that is used to treat autoimmune conditions, including multiple sclerosis and psoriasis. The mechanism of action of DMF is unclear but may involve the covalent modification of proteins or DMF serving as a prodrug that is converted to monomethyl fumarate (MMF). We found that DMF, but not MMF, blocked the activation of primary human and mouse T cells. Using a quantitative, site-specific chemical proteomic platform, we determined the DMF sensitivity of >2400 cysteine residues in human T cells. Cysteines sensitive to DMF, but not MMF, were identified in several proteins with established biochemical or genetic links to T cell function, including protein kinase Ctheta (PKCtheta). DMF blocked the association of PKCtheta with the costimulatory receptor CD28 by perturbing a CXXC motif in the C2 domain of this kinase. Mutation of these DMF-sensitive cysteines also impaired PKCtheta-CD28 interactions and T cell activation, designating the C2 domain of PKCtheta as a key functional, electrophile-sensing module important for T cell biology.
-
Edwards, L. J., et al (2015). "Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice" Clin Immunol 158(2): 221-230.
PubMed
The transcription factor STAT3 is overexpressed and hyperactivated in T cells from SLE patients. STAT3 plays a central role in T cell differentiation into Th17 and T follicular helper cells, two subsets that orchestrate autoimmune responses in SLE. Moreover, STAT3 is important in chemokine-mediated T cell migration. To better understand its role in SLE, we inhibited STAT3 in lupus-prone mice using the small molecule Stattic. Stattic-treated mice exhibited delayed onset of proteinuria (3 weeks later than controls), and had lower levels of anti-dsDNA antibodies and inflammatory cytokines. Inhibitor treatment reduced lymphadenopathy, resulted in a 3-fold decrease in total T cell number, and a 4-fold decrease in the numbers of T follicular helper cells. In vitro experiments showed that Stattic-treated T cells exhibited decreased proliferation and a decrease in ability to migrate to CXCL12. We propose that STAT3 inhibition represents a therapeutic target in SLE, particularly lupus nephritis.
-
Oh, Y. M., et al (2015). "Ndrg1 is a T-cell clonal anergy factor negatively regulated by CD28 costimulation and interleukin-2" Nat Commun 6: 8698.
PubMed
Induction of T-cell clonal anergy involves serial activation of transcription factors, including NFAT and Egr2/3. However, downstream effector mechanisms of these transcription factors are not fully understood yet. Here we identify Ndrg1 as an anergy factor induced by Egr2. Ndrg1 is upregulated by anergic signalling and maintained at high levels in resting anergic T cells. Overexpression of Ndrg1 mimics the anergic state and knockout of the gene prevents anergy induction. Interestingly, Ndrg1 is phosphorylated and degraded by CD28 signalling in a proteasome-dependent manner, explaining the costimulation dependence of anergy prevention. Similarly, IL-2 treatment of anergic T cells, under conditions that lead to the reversal of anergy, also induces Ndrg1 phosphorylation and degradation. Finally, older Ndrg1-deficient mice show T-cell hyperresponsiveness and Ndrg1-deficient T cells aggravate inducible autoimmune inflammation. Thus, Ndrg1 contributes to the maintenance of clonal anergy and inhibition of T-cell-mediated inflammation.
Product Citations
-
Inflammatory arthritis irAE may represent a unique autoimmune disease primarily driven by T cells but likely not autoantibodies.
In Sci Adv on 3 April 2026 by Zhu, X., Yu, Y., et al.
PubMed
The underlying immunopathogenesis of inflammatory arthritis (IA) immune-related adverse event (irAE) remains obscure. Unlike rheumatoid arthritis (RA), where autoantibodies and B cell dysfunction are central features, the contribution of humoral immunity to IA-irAE is unclear. Here, we performed immunophenotyping of peripheral blood from patients with IA-irAE and compared them with patients with seronegative RA, immune checkpoint inhibition-treated patients without irAE, and healthy controls. IA-irAE was marked with increased cytotoxic gene expression and metabolic activation in T cells and reduced CXCR3 and CCR6 expression in CD4+ T cells. Contrary to seronegative RA, patients with IA-irAE displayed no substantial elevation in autoantibody levels or atypical CD11c+CD21- B cells. IA-irAE was further characterized by elevated levels of interleukin-6 (IL-6), IL-12, and type I interferon, which correlated with the T cell activation phenotypes. Together, our findings define IA-irAE as a disease with certain immunological features distinctive from RA, representing a potentially T cell-driven, autoantibody-independent autoimmunity. These results offer insights into immune tolerance breakdown and therapeutic targeting in irAEs.
-
Inflammatory arthritis immune related adverse events represent a unique autoimmune disease entity primarily driven by T cells, but likely not autoantibodies
In medRxiv on 6 June 2025 by Zhu, X., Yu, Y., et al.
-
Polysialic acid is upregulated on activated immune cells and negatively regulates anticancer immune activity.
In Front Oncol on 4 April 2025 by Drummond-Guy, O., Daly, J., et al.
PubMed
Suppression of anticancer immune function is a key driver of tumorigenesis. Identifying molecular pathways that inhibit anticancer immunity is critical for developing novel immunotherapeutics. One such molecule that has recently been identified is the carbohydrate polysialic acid (polySia), whose expression is dramatically upregulated on both cancer cells and immune cells in breast cancer patient tissues. The role of polySia in the anticancer immune response, however, remains incompletely understood. In this study, we profile polySia expression on both healthy primary immune cells and on infiltrating immune cells in the tumour microenvironment (TME). These studies reveal polySia expression on multiple immune cell subsets in patient breast tumors. We find that stimulation of primary T-cells and macrophages in vitro induces a significant upregulation of polySia expression. We subsequently show that polySia is appended to a range of different carrier proteins within these immune cells. Finally, we find that selective removal of polySia can significantly potentiate killing of breast cancer cells by innate immune cells. These studies implicate polySia as a significant negative regulator of anticancer immunity.
-
The role of Pim-1 kinases in inflammatory signaling pathways.
In Inflamm Res on 1 October 2024 by Baek, H. S., Kim, N., et al.
PubMed
This observational study investigated the regulatory mechanism of Pim-1 in inflammatory signaling pathways.