InVivoPlus anti-mouse CD3ε
Product Details
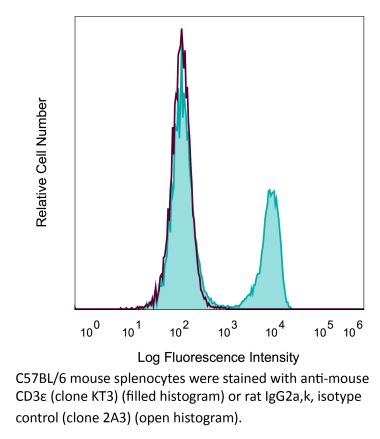
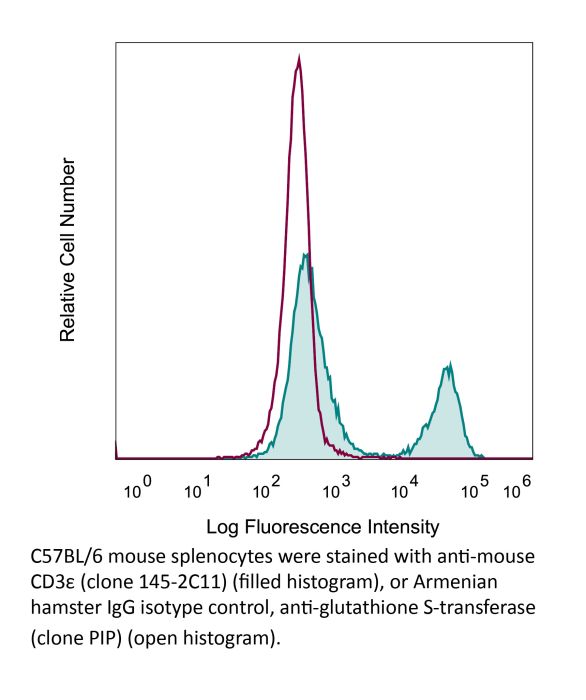
The 145-2C11 monoclonal antibody reacts with mouse CD3ε, a 20 kDa transmembrane cell-surface protein that belongs to the immunoglobulin superfamily. CD3ε is one of five polypeptide chains that combine to form the TCR complex. CD3ε is expressed on T lymphocytes, NK-T cells, and to varying degrees on developing thymocytes. CD3 plays roles in TCR signaling, T lymphocyte activation, and antigen recognition. The 145-2C11 antibody has been shown to induce T lymphocyte activation, proliferation, and apoptosis via binding and stimulating the TCR. Additionally, the 145-2C11 antibody has been reported to block the binding of the 17A2 antibody to CD3ε+ T lymphocytes.Specifications
| Isotype | Armenian Hamster IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse BM10-37 cytotoxic T cells |
| Reported Applications |
in vivo T cell depletion in vitro T cell stimulation/activation Immunofluorescence Flow cytometry Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_1107634 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus polyclonal Armenian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo T cell depletion
Glasner, A., et al. (2018). "NKp46 Receptor-Mediated Interferon-gamma Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis" Immunity 48(1): 107-119 e104. PubMed
Natural killer (NK) cells are innate lymphoid cells, and their presence within human tumors correlates with better prognosis. However, the mechanisms by which NK cells control tumors in vivo are unclear. Here, we used reflectance confocal microscopy (RCM) imaging in humans and in mice to visualize tumor architecture in vivo. We demonstrated that signaling via the NK cell receptor NKp46 (human) and Ncr1 (mouse) induced interferon-gamma (IFN-gamma) secretion from intratumoral NK cells. NKp46- and Ncr1-mediated IFN-gamma production led to the increased expression of the extracellular matrix protein fibronectin 1 (FN1) in the tumors, which altered primary tumor architecture and resulted in decreased metastases formation. Injection of IFN-gamma into tumor-bearing mice or transgenic overexpression of Ncr1 in NK cells in mice resulted in decreased metastasis formation. Thus, we have defined a mechanism of NK cell-mediated control of metastases in vivo that may help develop NK cell-dependent cancer therapies.
in vitro T cell stimulation/activation
Wendland, K., et al. (2018). "Retinoic Acid Signaling in Thymic Epithelial Cells Regulates Thymopoiesis" J Immunol 201(2): 524-532. PubMed
Despite the essential role of thymic epithelial cells (TEC) in T cell development, the signals regulating TEC differentiation and homeostasis remain incompletely understood. In this study, we show a key in vivo role for the vitamin A metabolite, retinoic acid (RA), in TEC homeostasis. In the absence of RA signaling in TEC, cortical TEC (cTEC) and CD80(lo)MHC class II(lo) medullary TEC displayed subset-specific alterations in gene expression, which in cTEC included genes involved in epithelial proliferation, development, and differentiation. Mice whose TEC were unable to respond to RA showed increased cTEC proliferation, an accumulation of stem cell Ag-1(hi) cTEC, and, in early life, a decrease in medullary TEC numbers. These alterations resulted in reduced thymic cellularity in early life, a reduction in CD4 single-positive and CD8 single-positive numbers in both young and adult mice, and enhanced peripheral CD8(+) T cell survival upon TCR stimulation. Collectively, our results identify RA as a regulator of TEC homeostasis that is essential for TEC function and normal thymopoiesis.
in vitro T cell stimulation/activation
Lacher, S. M., et al. (2018). "NF-kappaB inducing kinase (NIK) is an essential post-transcriptional regulator of T-cell activation affecting F-actin dynamics and TCR signaling" J Autoimmun 94: 110-121. PubMed
NF-kappaB inducing kinase (NIK) is the key protein of the non-canonical NF-kappaB pathway and is important for the development of lymph nodes and other secondary immune organs. We elucidated the specific role of NIK in T cells using T-cell specific NIK-deficient (NIK(DeltaT)) mice. Despite showing normal development of lymphoid organs, NIK(DeltaT) mice were resistant to induction of CNS autoimmunity. T cells from NIK(DeltaT) mice were deficient in late priming, failed to up-regulate T-bet and to transmigrate into the CNS. Proteomic analysis of activated NIK(-/-) T cells showed de-regulated expression of proteins involved in the formation of the immunological synapse: in particular, proteins involved in cytoskeleton dynamics. In line with this we found that NIK-deficient T cells were hampered in phosphorylation of Zap70, LAT, AKT, ERK1/2 and PLCgamma upon TCR engagement. Hence, our data disclose a hitherto unknown function of NIK in T-cell priming and differentiation.
in vitro T cell stimulation/activation
Ron-Harel, N., et al. (2016). "Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation" Cell Metab 24(1): 104-117. PubMed
Naive T cell stimulation activates anabolic metabolism to fuel the transition from quiescence to growth and proliferation. Here we show that naive CD4(+) T cell activation induces a unique program of mitochondrial biogenesis and remodeling. Using mass spectrometry, we quantified protein dynamics during T cell activation. We identified substantial remodeling of the mitochondrial proteome over the first 24 hr of T cell activation to generate mitochondria with a distinct metabolic signature, with one-carbon metabolism as the most induced pathway. Salvage pathways and mitochondrial one-carbon metabolism, fed by serine, contribute to purine and thymidine synthesis to enable T cell proliferation and survival. Genetic inhibition of the mitochondrial serine catabolic enzyme SHMT2 impaired T cell survival in culture and antigen-specific T cell abundance in vivo. Thus, during T cell activation, mitochondrial proteome remodeling generates specialized mitochondria with enhanced one-carbon metabolism that is critical for T cell activation and survival.
in vitro T cell stimulation/activation
Liu, H., et al. (2015). "The Immune Adaptor SLP-76 Binds to SUMO-RANGAP1 at Nuclear Pore Complex Filaments to Regulate Nuclear Import of Transcription Factors in T Cells" Mol Cell 59(5): 840-849. PubMed
While immune cell adaptors regulate proximal T cell signaling, direct regulation of the nuclear pore complex (NPC) has not been reported. NPC has cytoplasmic filaments composed of RanGAP1 and RanBP2 with the potential to interact with cytoplasmic mediators. Here, we show that the immune cell adaptor SLP-76 binds directly to SUMO-RanGAP1 of cytoplasmic fibrils of the NPC, and that this interaction is needed for optimal NFATc1 and NF-kappaB p65 nuclear entry in T cells. Transmission electron microscopy showed anti-SLP-76 cytoplasmic labeling of the majority of NPCs in anti-CD3 activated T cells. Further, SUMO-RanGAP1 bound to the N-terminal lysine 56 of SLP-76 where the interaction was needed for optimal RanGAP1-NPC localization and GAP exchange activity. While the SLP-76-RanGAP1 (K56E) mutant had no effect on proximal signaling, it impaired NF-ATc1 and p65/RelA nuclear entry and in vivo responses to OVA peptide. Overall, we have identified SLP-76 as a direct regulator of nuclear pore function in T cells.
in vitro T cell stimulation/activation
Xu, H., et al. (2015). "Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8" Nat Immunol . PubMed
Upon recognition of antigen, B cells undertake a bifurcated response in which some cells rapidly differentiate into plasmablasts while others undergo affinity maturation in germinal centers (GCs). Here we identified a double-negative feedback loop between the transcription factors IRF4 and IRF8 that regulated the initial developmental bifurcation of activated B cells as well as the GC response. IRF8 dampened signaling via the B cell antigen receptor (BCR), facilitated antigen-specific interaction with helper T cells, and promoted antibody affinity maturation while antagonizing IRF4-driven differentiation of plasmablasts. Genomic analysis revealed concentration-dependent actions of IRF4 and IRF8 in regulating distinct gene-expression programs. Stochastic modeling suggested that the double-negative feedback was sufficient to initiate bifurcation of the B cell developmental trajectories.
in vitro T cell stimulation/activation, Immunofluorescence
Kim, Y. U., et al. (2015). "Regulation of autoimmune germinal center reactions in lupus-prone BXD2 mice by follicular helper T cells" PLoS One 10(3): e0120294. PubMed
BXD2 mice spontaneously develop autoantibodies and subsequent glomerulonephritis, offering a useful animal model to study autoimmune lupus. Although initial studies showed a critical contribution of IL-17 and Th17 cells in mediating autoimmune B cell responses in BXD2 mice, the role of follicular helper T (Tfh) cells remains incompletely understood. We found that both the frequency of Th17 cells and the levels of IL-17 in circulation in BXD2 mice were comparable to those of wild-type. By contrast, the frequency of PD-1+ CXCR5+ Tfh cells was significantly increased in BXD2 mice compared with wild-type mice, while the frequency of PD-1+ CXCR5+ Foxp3+ follicular regulatory T (Tfr) cells was reduced in the former group. The frequency of Tfh cells rather than that of Th17 cells was positively correlated with the frequency of germinal center B cells as well as the levels of autoantibodies to dsDNA. More importantly, CXCR5+ CD4+ T cells isolated from BXD2 mice induced the production of IgG from naive B cells in an IL-21-dependent manner, while CCR6+ CD4+ T cells failed to do so. These results together demonstrate that Tfh cells rather than Th17 cells contribute to the autoimmune germinal center reactions in BXD2 mice.
in vitro T cell stimulation/activation
Awe, O., et al. (2015). "PU.1 Expression in T Follicular Helper Cells Limits CD40L-Dependent Germinal Center B Cell Development" J Immunol . PubMed
PU.1 is an ETS family transcription factor that is important for the development of multiple hematopoietic cell lineages. Previous work demonstrated a critical role for PU.1 in promoting Th9 development and in limiting Th2 cytokine production. Whether PU.1 has functions in other Th lineages is not clear. In this study, we examined the effects of ectopic expression of PU.1 in CD4+ T cells and observed decreased expression of genes involved with the function of T follicular helper (Tfh) cells, including Il21 and Tnfsf5 (encoding CD40L). T cells from conditional mutant mice that lack expression of PU.1 in T cells (Sfpi1lck-/-) demonstrated increased production of CD40L and IL-21 in vitro. Following adjuvant-dependent or adjuvant-independent immunization, we observed that Sfpi1lck-/- mice had increased numbers of Tfh cells, increased germinal center B cells (GCB cells), and increased Ab production in vivo. This correlated with increased expression of IL-21 and CD40L in Tfh cells from Sfpi1lck-/- mice compared with control mice. Finally, although blockade of IL-21 did not affect GCB cells in Sfpi1lck-/- mice, anti-CD40L treatment of immunized Sfpi1lck-/- mice decreased GCB cell numbers and Ag-specific Ig concentrations. Together, these data indicate an inhibitory role for PU.1 in the function of Tfh cells, germinal centers, and Tfh-dependent humoral immunity.
in vitro T cell stimulation/activation
Huang, Y., et al. (2015). "CRK proteins selectively regulate T cell migration into inflamed tissues" J Clin Invest 125(3): 1019-1032. PubMed
Effector T cell migration into inflamed sites greatly exacerbates tissue destruction and disease severity in inflammatory diseases, including graft-versus-host disease (GVHD). T cell migration into such sites depends heavily on regulated adhesion and migration, but the signaling pathways that coordinate these functions downstream of chemokine receptors are largely unknown. Using conditional knockout mice, we found that T cells lacking the adaptor proteins CRK and CRK-like (CRKL) exhibit reduced integrin-dependent adhesion, chemotaxis, and diapedesis. Moreover, these two closely related proteins exhibited substantial functional redundancy, as ectopic expression of either protein rescued defects in T cells lacking both CRK and CRKL. We determined that CRK proteins coordinate with the RAP guanine nucleotide exchange factor C3G and the adhesion docking molecule CASL to activate the integrin regulatory GTPase RAP1. CRK proteins were required for effector T cell trafficking into sites of inflammation, but not for migration to lymphoid organs. In a murine bone marrow transplantation model, the differential migration of CRK/CRKL-deficient T cells resulted in efficient graft-versus-leukemia responses with minimal GVHD. Together, the results from our studies show that CRK family proteins selectively regulate T cell adhesion and migration at effector sites and suggest that these proteins have potential as therapeutic targets for preventing GVHD.
in vitro T cell stimulation/activation
Gu, A. D., et al. (2015). "A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling" Immunity 42(1): 68-79. PubMed
Transforming growth factor-beta (TGF-beta) suppresses T cell function to maintain self-tolerance and to promote tumor immune evasion. Yet how Smad4, a transcription factor component of TGF-beta signaling, regulates T cell function remains unclear. Here we have demonstrated an essential role for Smad4 in promoting T cell function during autoimmunity and anti-tumor immunity. Smad4 deletion rescued the lethal autoimmunity resulting from transforming growth factor-beta receptor (TGF-betaR) deletion and compromised T-cell-mediated tumor rejection. Although Smad4 was dispensable for T cell generation, homeostasis, and effector function, it was essential for T cell proliferation after activation in vitro and in vivo. The transcription factor Myc was identified to mediate Smad4-controlled T cell proliferation. This study thus reveals a requirement of Smad4 for T-cell-mediated autoimmunity and tumor rejection, which is beyond the current paradigm. It highlights a TGF-betaR-independent role for Smad4 in promoting T cell function, autoimmunity, and anti-tumor immunity.
in vitro T cell stimulation/activation
Rabenstein, H., et al. (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517. PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vitro T cell stimulation/activation
Bertin, S., et al. (2014). "The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells" Nat Immunol 15(11): 1055-1063. PubMed
TRPV1 is a Ca(2+)-permeable channel studied mostly as a pain receptor in sensory neurons. However, its role in other cell types is poorly understood. Here we found that TRPV1 was functionally expressed in CD4(+) T cells, where it acted as a non-store-operated Ca(2+) channel and contributed to T cell antigen receptor (TCR)-induced Ca(2+) influx, TCR signaling and T cell activation. In models of T cell-mediated colitis, TRPV1 promoted colitogenic T cell responses and intestinal inflammation. Furthermore, genetic and pharmacological inhibition of TRPV1 in human CD4(+) T cells recapitulated the phenotype of mouse Trpv1(-/-) CD4(+) T cells. Our findings suggest that inhibition of TRPV1 could represent a new therapeutic strategy for restraining proinflammatory T cell responses.
in vitro T cell stimulation/activation, Flow Cytometry
Tang, W., et al. (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566. PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
in vitro T cell stimulation/activation
Vegran, F., et al. (2014). "The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells" Nat Immunol 15(8): 758-766. PubMed
The TH9 subset of helper T cells was initially shown to contribute to the induction of autoimmune and allergic diseases, but subsequent evidence has suggested that these cells also exert antitumor activities. However, the molecular events that account for their effector properties are elusive. Here we found that the transcription factor IRF1 enhanced the effector function of TH9 cells and dictated their anticancer properties. Under TH9-skewing conditions, interleukin 1beta (IL-1beta) induced phosphorylation of the transcription factor STAT1 and subsequent expression of IRF1, which bound to the promoters of Il9 and Il21 and enhanced secretion of the cytokines IL-9 and IL-21 from TH9 cells. Furthermore, IL-1beta-induced TH9 cells exerted potent anticancer functions in an IRF1- and IL-21-dependent manner. Our findings thus identify IRF1 as a target for controlling the function of TH9 cells.
in vitro T cell stimulation/activation
Berger, H., et al. (2013). "SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth" Cancer Res 73(12): 3578-3590. PubMed
Activation of the transcription factor PPARgamma by the n-3 fatty acid docosahexaenoic acid (DHA) is implicated in controlling proinflammatory cytokine secretion, but the intracellular signaling pathways engaged by PPARgamma are incompletely characterized. Here, we identify the adapter-encoding gene SOCS3 as a critical transcriptional target of PPARgamma. SOCS3 promoter binding and gene transactivation by PPARgamma was associated with a repression in differentiation of proinflammatory T-helper (TH)17 cells. Accordingly, TH17 cells induced in vitro displayed increased SOCS3 expression and diminished capacity to produce interleukin (IL)-17 following activation of PPARgamma by DHA. Furthermore, naive CD4 T cells derived from mice fed a DHA-enriched diet displayed less capability to differentiate into TH17 cells. In two different mouse models of cancer, DHA prevented tumor outgrowth and angiogenesis in an IL-17-dependent manner. Altogether, our results uncover a novel molecular pathway by which PPARgamma-induced SOCS3 expression prevents IL-17-mediated cancer growth.
in vitro T cell stimulation/activation
Sledzinska, A., et al. (2013). "TGF-beta signalling is required for CD4(+) T cell homeostasis but dispensable for regulatory T cell function" PLoS Biol 11(10): e1001674. PubMed
TGF-beta is widely held to be critical for the maintenance and function of regulatory T (T(reg)) cells and thus peripheral tolerance. This is highlighted by constitutive ablation of TGF-beta receptor (TR) during thymic development in mice, which leads to a lethal autoimmune syndrome. Here we describe that TGF-beta-driven peripheral tolerance is not regulated by TGF-beta signalling on mature CD4(+) T cells. Inducible TR2 ablation specifically on CD4(+) T cells did not result in a lethal autoinflammation. Transfer of these TR2-deficient CD4(+) T cells to lymphopenic recipients resulted in colitis, but not overt autoimmunity. In contrast, thymic ablation of TR2 in combination with lymphopenia led to lethal multi-organ inflammation. Interestingly, deletion of TR2 on mature CD4(+) T cells does not result in the collapse of the T(reg) cell population as observed in constitutive models. Instead, a pronounced enlargement of both regulatory and effector memory T cell pools was observed. This expansion is cell-intrinsic and seems to be caused by increased T cell receptor sensitivity independently of common gamma chain-dependent cytokine signals. The expression of Foxp3 and other regulatory T cells markers was not dependent on TGF-beta signalling and the TR2-deficient T(reg) cells retained their suppressive function both in vitro and in vivo. In summary, absence of TGF-beta signalling on mature CD4(+) T cells is not responsible for breakdown of peripheral tolerance, but rather controls homeostasis of mature T cells in adult mice.
in vitro T cell stimulation/activation
Goswami, R., et al. (2012). "STAT6-dependent regulation of Th9 development" J Immunol 188(3): 968-975. PubMed
Th cell effector subsets develop in response to specific cytokine environments. The development of a particular cytokine-secreting pattern requires an integration of signals that may promote the development of opposing pathways. A recent example of this paradigm is the IL-9-secreting Th9 cell that develops in response to TGF-beta and IL-4, cytokines that, in isolation, promote the development of inducible regulatory T cells and Th2 cells, respectively. To determine how the balance of these factors results in priming for IL-9 secretion, we examined the effects of each pathway on transcription factors that regulate Th cell differentiation. We demonstrated that TGF-beta induces the PU.1-encoding Sfpi1 locus and that this is independent of IL-4-induced STAT6 activation. IL-4-activated STAT6 is required for repressing the expression of T-bet and Foxp3 in Th9 cells, transcription factors that inhibit IL-9 production, and STAT6 is required for the induction of IRF4, which promotes Th9 development. These data established a transcription factor network that regulates IL-9 and demonstrated how combinations of cytokine signals generate cytokine-secreting potential by altering the expression of a panel of transcription factors.
in vivo T cell depletion
Peng, B., et al. (2009). "Anti-CD3 antibodies modulate anti-factor VIII immune responses in hemophilia A mice after factor VIII plasmid-mediated gene therapy" Blood 114(20): 4373-4382. PubMed
One major obstacle in gene therapy is the generation of immune responses directed against transgene product. Five consecutive anti-CD3 treatments concomitant with factor VIII (FVIII) plasmid injection prevented the formation of inhibitory antibodies against FVIII and achieved persistent, therapeutic levels of FVIII gene expression in treated hemophilia A mice. Repeated plasmid gene transfer is applicable in tolerized mice without eliciting immune responses. Anti-CD3 treatment significantly depleted both CD4+ and CD8+ T cells, whereas increased transforming growth factor-beta levels in plasma and the frequency of both CD4+CD25+FoxP3+ and CD4+CD25-Foxp3+ regulatory T cells in the initial few weeks after treatment. Although prior depletion of CD4+CD25+ cells did not abrogate tolerance induction, adoptive transfer of CD4+ cells from tolerized mice at 6 weeks after treatment protected recipient mice from anti-FVIII immune responses. Anti-CD3-treated mice mounted immune responses against both T-dependent and T-independent neo-antigens, indicating that anti-CD3 did not hamper the immune systems in the long term. Concomitant FVIII plasmid + anti-CD3 treatment induced long-term tolerance specific to FVIII via a mechanism involving the increase in transforming growth factor-beta levels and the generation of adaptive FVIII-specific CD4+Foxp3+ regulatory T cells at the periphery. Furthermore, anti-CD3 can reduce the titers of preexisting anti-FVIII inhibitory antibodies in hemophilia A mice.
in vitro T cell stimulation/activation
Dardalhon, V., et al. (2008). "IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells" Nat Immunol 9(12): 1347-1355. PubMed
Transcription factor Foxp3 is critical for generating regulatory T cells (T(reg) cells). Transforming growth factor-beta (TGF-beta) induces Foxp3 and suppressive T(reg) cells from naive T cells, whereas interleukin 6 (IL-6) inhibits the generation of inducible T(reg) cells. Here we show that IL-4 blocked the generation of TGF-beta-induced Foxp3(+) T(reg) cells and instead induced a population of T helper cells that produced IL-9 and IL-10. The IL-9(+)IL-10(+) T cells demonstrated no regulatory properties despite producing abundant IL-10. Adoptive transfer of IL-9(+)IL-10(+) T cells into recombination-activating gene 1-deficient mice induced colitis and peripheral neuritis, the severity of which was aggravated if the IL-9(+)IL-10(+) T cells were transferred with CD45RB(hi) CD4(+) effector T cells. Thus IL-9(+)IL-10(+) T cells lack suppressive function and constitute a distinct population of helper-effector T cells that promote tissue inflammation.
- Immunology and Microbiology,
Induction of immortal-like and functional CAR T cells by defined factors.
In The Journal of Experimental Medicine on 6 May 2024 by Wang, L., Jin, G., et al.
PubMed
Long-term antitumor efficacy of chimeric antigen receptor (CAR) T cells depends on their functional persistence in vivo. T cells with stem-like properties show better persistence, but factors conferring bona fide stemness to T cells remain to be determined. Here, we demonstrate the induction of CAR T cells into an immortal-like and functional state, termed TIF. The induction of CARTIF cells depends on the repression of two factors, BCOR and ZC3H12A, and requires antigen or CAR tonic signaling. Reprogrammed CARTIF cells possess almost infinite stemness, similar to induced pluripotent stem cells while retaining the functionality of mature T cells, resulting in superior antitumor effects. Following the elimination of target cells, CARTIF cells enter a metabolically dormant state, persisting in vivo with a saturable niche and providing memory protection. TIF represents a novel state of T cells with unprecedented stemness, which confers long-term functional persistence of CAR T cells in vivo and holds broad potential in T cell therapies. © 2024 Wang et al.
- Mus musculus (House mouse),
- Immunology and Microbiology,
- Cell Biology,
- Biochemistry and Molecular biology
TMEM41B is an endoplasmic reticulum Ca2+release channel maintaining T cell metabolic quiescence and responsiveness
Preprint on BioRxiv : the Preprint Server for Biology on 26 December 2023 by Ma, Y., Wang, Y., et al.
PubMed
SUMMARY Naive T cells are metabolically quiescent. Here, we report an unexpected role of endoplasmic reticulum (ER) Ca 2+ in preserving T cell metabolic quiescence. TMEM41B, an ER-resident membrane protein previously known for its crucial roles in autophagy, lipid scrabbling and viral infections, is identified as a novel type of concentration-dependent ER Ca 2+ release channel. Ablation of TMEM41B induces ER Ca 2+ overload, triggering the upregulation of IL-2 and IL-7 receptors in naive T cells. Consequently, this leads to increased basal signaling of the JAK-STAT, AKT-mTOR, and MAPK pathways, propelling TMEM41B-deficient naive T cells into a metabolically activated yet immunologically naive state. ER Ca 2+ overload also downregulates CD5, a suppressor of TCR signaling, thereby reducing the activation threshold of TMEM41B-deficient T cells, resulting in attenuated tolerance and heightened T cell responses during infections. In summary, TMEM41B-mediated ER Ca 2+ release is a pivotal determinant governing metabolic quiescence and responsiveness of naive T cells.
- Mus musculus (House mouse),
- Immunology and Microbiology
One infusion of engineered long-lasting and multifunctional T cells cures asthma in mice
Preprint on BioRxiv : the Preprint Server for Biology on 6 November 2023 by Jin, G., Liu, Y., et al.
PubMed
Summary The majority of common chronic human diseases are incurable, which affect a large portion of population and require life-long treatments that impose huge disease, economic and social burden. For example, asthma is the most common respiratory disease affecting over 300 million people and accounts for > 250,000 death annually, for which there is no cure. Unlike traditional therapyies, engineered T cells, such as chimeric antigen receptor CAR T (CAR-T) cells, function as living drugs and can cure some hematological malignancies, but whether engineered T cells can cure common diseases beyond cancer remains elusive. Here, we develop a curative therapy for asthma based on enginerred T cell. With IL-5 as the targeting domain and depletion of BCOR and ZC3H12A, we produce long-lasting CAR-T cells eradicating IL-5Rα + eosinophils, termed I mmortal-like and F unctional IL-5 CAR-T cells (5T IF ) cells. We further enginerred 5T IF cells to secrete an IL-4 mutein that blocks the singaling of both IL-4 and IL-13, two driver inflammatory cytokines in asthma, named as 5T IF 4 cells. In multiple models of asthma, one infusion of 5T IF 4 cells in fully immunocompetent mice, in the absence of any conditioning regimen, confers long-term depletion of pathological eosinophils and blockade of IL-4/IL-13 actions, resulting in sustained repression of type 2 inflammation and asthmatic symptoms. Furthurmore, 5T IF 4 cells can also be induced in human T cells in NSG mice. Our data demonstrate that asthma, a common noncancerous disease, can be cured by a single infusion of engineered long-lasting and multifunctional T cells, which paves the way for curing common chronic diseases by engineered long-lived T cells.
- Immunology and Microbiology
Induction of immortal-like and functional CAR-T cells by defined factors
Preprint on BioRxiv : the Preprint Server for Biology on 6 November 2023 by Wang, L., Jin, G., et al.
PubMed
Long-term antitumor efficacy of chimeric antigen receptor (CAR)-T cells depends on their functional persistence in vivo. T cells with stem-like property show better persistence, but factors conferring T cells bona fide stemness remain to be determined. Here, we demonstrate the induction of CAR-T cells into an immortal-like and functional state, termed TIF. The induction of CAR-TIF cells depends on repression of two factors: BCOR and ZC3H12A, and requires antigen or CAR tonic signaling. The reprogrammed CAR-TIF cells possess almost infinite stemness resembling induced pluripotent stem cells but retain functionality of mature T cells, which exhibit superior antitumor effect. CAR-TIF cells enter a metabolically dormant state after elimination of target cells, which persist in vivo with a saturable niche and mediate memory protection. CAR-TIF cells represent a novel state of T cells with unprecedented stemness conferring long-term functional persistence of CAR-T cells in vivo, which have broad potentials in T cell therapies.
- Mus musculus (House mouse),
- Immunology and Microbiology
An essential role for miR-15/16 in Treg suppression and restriction of proliferation.
In Cell Reports on 31 October 2023 by Johansson, K., Gagnon, J. D., et al.
PubMed
The miR-15/16 family targets a large network of genes in T cells to restrict their cell cycle, memory formation, and survival. Upon T cell activation, miR-15/16 are downregulated, allowing rapid expansion of differentiated effector T cells to mediate a sustained response. Here, we used conditional deletion of miR-15/16 in regulatory T cells (Tregs) to identify immune functions of the miR-15/16 family in T cells. miR-15/16 are indispensable to maintain peripheral tolerance by securing efficient suppression by a limited number of Tregs. miR-15/16 deficiency alters expression of critical Treg proteins and results in accumulation of functionally impaired FOXP3loCD25loCD127hi Tregs. Excessive proliferation in the absence of miR-15/16 shifts Treg fate and produces an effector Treg phenotype. These Tregs fail to control immune activation, leading to spontaneous multi-organ inflammation and increased allergic inflammation in a mouse model of asthma. Together, our results demonstrate that miR-15/16 expression in Tregs is essential to maintain immune tolerance. Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
- Mus musculus (House mouse),
- Cancer Research,
- Immunology and Microbiology,
- Cell Biology,
- Biochemistry and Molecular biology
Molecular, metabolic, and functional CD4 T cell paralysis in the lymph node impedes tumor control.
In Cell Reports on 26 September 2023 by Guo, M., Abd-Rabbo, D., et al.
PubMed
CD4 T cells are central effectors of anti-cancer immunity and immunotherapy, yet the regulation of CD4 tumor-specific T (TTS) cells is unclear. We demonstrate that CD4 TTS cells are quickly primed and begin to divide following tumor initiation. However, unlike CD8 TTS cells or exhaustion programming, CD4 TTS cell proliferation is rapidly frozen in place by a functional interplay of regulatory T cells and CTLA4. Together these mechanisms paralyze CD4 TTS cell differentiation, redirecting metabolic circuits, and reducing their accumulation in the tumor. The paralyzed state is actively maintained throughout cancer progression and CD4 TTS cells rapidly resume proliferation and functional differentiation when the suppressive constraints are alleviated. Overcoming their paralysis established long-term tumor control, demonstrating the importance of rapidly crippling CD4 TTS cells for tumor progression and their potential restoration as therapeutic targets. Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
- In Vivo,
- Mus musculus (House mouse),
- Cancer Research
Tumor PD-L1 engages myeloid PD-1 to suppress type I interferon to impair cytotoxic T lymphocyte recruitment.
In Cancer Cell on 13 March 2023 by Klement, J. D., Redd, P. S., et al.
PubMed
The cellular and molecular mechanisms underlying tumor cell PD-L1 (tPD-L1) function in tumor immune evasion are incompletely understood. We report here that tPD-L1 does not suppress cytotoxic T lymphocyte (CTL) activity in co-cultures of tumor cells and tumor-specific CTLs and exhibits no effect on primary tumor growth. However, deleting tPD-L1 decreases lung metastasis in a CTL-dependent manner in tumor-bearing mice. Depletion of myeloid cells or knocking out PD-1 in myeloid cells (mPD-1) impairs tPD-L1 promotion of tumor lung metastasis in mice. Single-cell RNA sequencing (scRNA-seq) reveals that tPD-L1 engages mPD-1 to activate SHP2 to antagonize the type I interferon (IFN-I) and STAT1 pathway to repress Cxcl9 and impair CTL recruitment to lung metastases. Human cancer patient response to PD-1 blockade immunotherapy correlates with IFN-I response in myeloid cells. Our findings determine that tPD-L1 engages mPD-1 to activate SHP2 to suppress the IFN-I-STAT1-CXCL9 pathway to impair CTL tumor recruitment in lung metastasis. Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
- Genetics,
- Immunology and Microbiology
Forced expression of the non-coding RNA miR-17∼92 restores activation and function in CD28-deficient CD4+ T cells.
In IScience on 18 November 2022 by Dölz, M., Hasiuk, M., et al.
PubMed
CD28 provides the prototypical costimulatory signal required for productive T-cell activation. Known molecular consequences of CD28 costimulation are mostly based on studies of protein signaling molecules. The microRNA cluster miR-17∼92 is induced by T cell receptor stimulation and further enhanced by combined CD28 costimulation. We demonstrate that transgenic miR-17∼92 cell-intrinsically largely overcomes defects caused by CD28 deficiency. Combining genetics, transcriptomics, bioinformatics, and biochemical miRNA:mRNA interaction maps we empirically validate miR-17∼92 target genes that include several negative regulators of T cell activation. CD28-deficient T cells exhibit derepressed miR-17∼92 target genes during activation. CRISPR/Cas9-mediated ablation of the miR-17∼92 targets Pten and Nrbp1 in naive CD28-/- CD4+ T cells differentially increases proliferation and expression of the activation markers CD25 and CD44, respectively. Thus, we propose that miR-17∼92 constitutes a central mediator for T cell activation, integrating signals by the TCR and CD28 costimulation by dampening multiple brakes that prevent T cell activation. © 2022 The Author(s).
- Mus musculus (House mouse),
- Biochemistry and Molecular biology,
- Immunology and Microbiology
Transcription factor RORα enforces stability of the Th17 cell effector program by binding to a Rorc cis-regulatory element.
In Immunity on 8 November 2022 by Hall, J. A., Pokrovskii, M., et al.
PubMed
T helper 17 (Th17) cells regulate mucosal barrier defenses but also promote multiple autoinflammatory diseases. Although many molecular determinants of Th17 cell differentiation have been elucidated, the transcriptional programs that sustain Th17 cells in vivo remain obscure. The transcription factor RORγt is critical for Th17 cell differentiation; however, it is not clear whether the closely related RORα, which is co-expressed in Th17 cells, has a distinct role. Here, we demonstrated that although dispensable for Th17 cell differentiation, RORα was necessary for optimal Th17 responses in peripheral tissues. The absence of RORα in T cells led to reductions in both RORγt expression and effector function among Th17 cells. Cooperative binding of RORα and RORγt to a previously unidentified Rorc cis-regulatory element was essential for Th17 lineage maintenance in vivo. These data point to a non-redundant role of RORα in Th17 lineage maintenance via reinforcement of the RORγt transcriptional program.Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
- Mus musculus (House mouse),
- Immunology and Microbiology
Identification of environmental factors that promote intestinal inflammation.
In Nature on 1 November 2022 by Sanmarco, L. M., Chao, C. C., et al.
PubMed
Genome-wide association studies have identified risk loci linked to inflammatory bowel disease (IBD)1-a complex chronic inflammatory disorder of the gastrointestinal tract. The increasing prevalence of IBD in industrialized countries and the augmented disease risk observed in migrants who move into areas of higher disease prevalence suggest that environmental factors are also important determinants of IBD susceptibility and severity2. However, the identification of environmental factors relevant to IBD and the mechanisms by which they influence disease has been hampered by the lack of platforms for their systematic investigation. Here we describe an integrated systems approach, combining publicly available databases, zebrafish chemical screens, machine learning and mouse preclinical models to identify environmental factors that control intestinal inflammation. This approach established that the herbicide propyzamide increases inflammation in the small and large intestine. Moreover, we show that an AHR-NF-κB-C/EBPβ signalling axis operates in T cells and dendritic cells to promote intestinal inflammation, and is targeted by propyzamide. In conclusion, we developed a pipeline for the identification of environmental factors and mechanisms of pathogenesis in IBD and, potentially, other inflammatory diseases. © 2022. The Author(s), under exclusive licence to Springer Nature Limited.
- Cancer Research,
- Immunology and Microbiology
Balance between immunoregulatory B cells and plasma cells drives pancreatic tumor immunity.
In Cell Reports Medicine on 20 September 2022 by Mirlekar, B., Wang, Y., et al.
PubMed
Plasma cell responses are associated with anti-tumor immunity and favorable response to immunotherapy. B cells can amplify anti-tumor immune responses through antibody production; yet B cells in patients and tumor-bearing mice often fail to support this effector function. We identify dysregulated transcriptional program in B cells that disrupts differentiation of naive B cells into anti-tumor plasma cells. The signaling network contributing to this dysfunction is driven by interleukin (IL) 35 stimulation of a STAT3-PAX5 complex that upregulates the transcriptional regulator BCL6 in naive B cells. Transient inhibition of BCL6 in tumor-educated naive B cells is sufficient to reverse the dysfunction in B cell differentiation, stimulating the intra-tumoral accumulation of plasma cells and effector T cells and rendering pancreatic tumors sensitive to anti-programmed cell death protein 1 (PD-1) blockade. Our findings argue that B cell effector dysfunction in cancer can be due to an active systemic suppression program that can be targeted to synergize with T cell-directed immunotherapy.Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
- FC/FACS,
- Mus musculus (House mouse),
- Immunology and Microbiology
Overexpressing Kallistatin Aggravates Experimental Autoimmune Uveitis Through Promoting Th17 Differentiation.
In Frontiers in Immunology on 5 November 2021 by Chen, N., Chen, S., et al.
PubMed
Kallistatin or kallikrein-binding protein (KBP) has been reported to regulate angiogenesis, inflammation and tumor progression. Autoimmune uveitis is a common, sight-threatening inflammatory intraocular disease. However, the roles of kallistatin in autoimmunity and autoreactive T cells are poorly investigated. Compared to non-uveitis controls, we found that plasma levels of kallistatin were significantly upregulated in patients with Vogt-Koyanagi-Harada (VKH) disease, one of the non-infectious uveitis. Using an experimental autoimmune uveitis (EAU) model induced by human interphotoreceptor retinoid-binding protein peptide 651-670 (hIRBP651-670), we examined the effects of kallistatin on the pathogenesis of autoimmune diseases. Compared to wild type (WT) mice, kallistatin transgenic (KS) mice developed severe uveitis with dominant Th17 infiltrates in the eye. In addition, the proliferative antigen-specific T cells isolated from KS EAU mice produced increased levels of IL-17A, but not IFN-γ or IL-10 cytokines. Moreover, splenic CD4+ T cells from naïve KS mice expressed higher levels of Il17a mRNA compared to WT naïve mice. Under Th17 polarization conditions, KS mice exhibited enhanced differentiation of naïve CD4+ T cells into Th17 cells compared to WT controls. Together, our results indicate that kallistatin promotes Th17 differentiation and is a key regulator of aggravating autoinflammation in EAU. Targeting kallistatin might be a potential to treat autoimmune disease. Copyright © 2021 Chen, Chen, Zhang, Cui, Wu, Guo, Shao, Ma and Zhang.
- Mus musculus (House mouse),
- Immunology and Microbiology
BCL6 controls contact-dependent help delivery during follicular T-B cell interactions.
In Immunity on 12 October 2021 by Liu, D., Yan, J., et al.
PubMed
BCL6 is required for development of follicular T helper (Tfh) cells to support germinal center (GC) formation. However, it is not clear what unique functions programmed by BCL6 can explain its absolute essentiality in T cells for GC formation. We found that ablation of one Bcl6 allele did not appreciably alter early T cell activation and follicular localization but inhibited GC formation and Tfh cell maintenance. BCL6 impinged on Tfh calcium signaling and also controlled Tfh entanglement with and CD40L delivery to B cells. Amounts of BCL6 protein and nominal frequencies of Tfh cells markedly changed within hours after strengths of T-B cell interactions were altered in vivo, while CD40L overexpression rectified both defective GC formation and Tfh cell maintenance because of the BCL6 haploinsufficiency. Our results reveal BCL6 functions in Tfh cells that are essential for GC formation and suggest that BCL6 helps maintain Tfh cell phenotypes in a T cell non-autonomous manner. Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
- Mus musculus (House mouse),
- Biochemistry and Molecular biology,
- Cell Biology
Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy.
In Cell on 6 August 2020 by Wu, L., Hollinshead, K. E. R., et al.
PubMed
Targeting glycolysis has been considered therapeutically intractable owing to its essential housekeeping role. However, the context-dependent requirement for individual glycolytic steps has not been fully explored. We show that CRISPR-mediated targeting of glycolysis in T cells in mice results in global loss of Th17 cells, whereas deficiency of the glycolytic enzyme glucose phosphate isomerase (Gpi1) selectively eliminates inflammatory encephalitogenic and colitogenic Th17 cells, without substantially affecting homeostatic microbiota-specific Th17 cells. In homeostatic Th17 cells, partial blockade of glycolysis upon Gpi1 inactivation was compensated by pentose phosphate pathway flux and increased mitochondrial respiration. In contrast, inflammatory Th17 cells experience a hypoxic microenvironment known to limit mitochondrial respiration, which is incompatible with loss of Gpi1. Our study suggests that inhibiting glycolysis by targeting Gpi1 could be an effective therapeutic strategy with minimum toxicity for Th17-mediated autoimmune diseases, and, more generally, that metabolic redundancies can be exploited for selective targeting of disease processes. Copyright © 2020 Elsevier Inc. All rights reserved.
- Cardiovascular biology
Contrast-enhanced ultrasound measurement of pancreatic blood flow dynamics predicts type1 diabetes therapeutic reversal in preclinical models
Preprint on BioRxiv : the Preprint Server for Biology on 5 March 2020 by Pham, V., Ramirez, D. G., et al.
PubMed
In type 1 diabetes (T1D) immune-cell infiltration into the islets of Langerhans (insulitis) and β-cell decline occurs many years before diabetes presents. Non-invasively detecting insulitis and β-cell decline would allow diagnosis of eventual diabetes and provide a means to monitor the efficacy of therapeutic intervention. However, there is a lack of validated clinical approaches for non-invasively imaging disease progression leading to T1D. Islets have a dense microvasculature that reorganizes during diabetes. We previously demonstrated contrast-enhanced ultrasound measurements of pancreatic blood-flow dynamics could predict disease progression in T1D pre-clinical models. Here we test whether these measurements can predict successful therapeutic prevention of T1D. We performed destruction-reperfusion measurements using a small-animal ultrasound machine and size-isolated microbubbles, in NOD-scid mice receiving an adoptive transfer of diabetogenic splenocytes (AT mice). Mice received vehicle control or either of the following treatments: 1) antiCD4 to deplete CD4 + T cells; 2) antiCD3 to block T cell activation, 3) Verapamil to reduce β-cell apoptosis and 4) TUDCA to reduce ER stress. We compared measurements of pancreas blood-flow dynamics with subsequent progression to diabetes. In AT mice blood-flow dynamics were altered >2 weeks after splenocyte transfer. AntiCD4, antiCD3 and verapamil provided a significant delay in diabetes development. Treated AT mice with delayed or absent diabetes development showed significantly altered blood flow dynamics compared to untreated AT mice. Conversely, treated AT mice that developed diabetes, despite therapy, showed similar blood-flow dynamics to untreated AT mice. Thus, contrast-enhanced ultrasound measurement of pancreas blood-flow dynamics can predict the successful or unsuccessful delay or prevention of diabetes upon therapeutic treatments that target both immune activity or β-cell protection. This strategy may provide a clinically deployable predictive marker for disease progression and therapeutic reversal in asymptomatic T1D.
- Mus musculus (House mouse),
- Biochemistry and Molecular biology,
- Cell Biology,
- Immunology and Microbiology
Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function.
In Cell Metabolism on 4 February 2020 by Field, C. S., Baixauli, F., et al.
PubMed
Regulatory T cells (Tregs) subdue immune responses. Central to Treg activation are changes in lipid metabolism that support their survival and function. Fatty acid binding proteins (FABPs) are a family of lipid chaperones required to facilitate uptake and intracellular lipid trafficking. One family member, FABP5, is expressed in T cells, but its function remains unclear. We show that in Tregs, genetic or pharmacologic inhibition of FABP5 function causes mitochondrial changes underscored by decreased OXPHOS, impaired lipid metabolism, and loss of cristae structure. FABP5 inhibition in Tregs triggers mtDNA release and consequent cGAS-STING-dependent type I IFN signaling, which induces heightened production of the regulatory cytokine IL-10 and promotes Treg suppressive activity. We find evidence of this pathway, along with correlative mitochondrial changes in tumor infiltrating Tregs, which may underlie enhanced immunosuppression in the tumor microenvironment. Together, our data reveal that FABP5 is a gatekeeper of mitochondrial integrity that modulates Treg function. Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
- Mus musculus (House mouse),
- Immunology and Microbiology
Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease.
In Cell on 9 January 2020 by Lee, J. Y., Hall, J. A., et al.
PubMed
Lymphoid cells that produce interleukin (IL)-17 cytokines protect barrier tissues from pathogenic microbes but are also prominent effectors of inflammation and autoimmune disease. T helper 17 (Th17) cells, defined by RORγt-dependent production of IL-17A and IL-17F, exert homeostatic functions in the gut upon microbiota-directed differentiation from naive CD4+ T cells. In the non-pathogenic setting, their cytokine production is regulated by serum amyloid A proteins (SAA1 and SAA2) secreted by adjacent intestinal epithelial cells. However, Th17 cell behaviors vary markedly according to their environment. Here, we show that SAAs additionally direct a pathogenic pro-inflammatory Th17 cell differentiation program, acting directly on T cells in collaboration with STAT3-activating cytokines. Using loss- and gain-of-function mouse models, we show that SAA1, SAA2, and SAA3 have distinct systemic and local functions in promoting Th17-mediated inflammatory diseases. These studies suggest that T cell signaling pathways modulated by the SAAs may be attractive targets for anti-inflammatory therapies. Copyright © 2019 Elsevier Inc. All rights reserved.
- Cell Biology
TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis.
In Nature Cell Biology on 1 December 2019 by Zhu, L., Li, Y., et al.
PubMed
TANK-binding kinase 1 (TBK1) responds to microbial stimuli and mediates the induction of type I interferon (IFN). Here, we show that TBK1 is also a central mediator of growth factor signalling; this function of TBK1 relies on a specific adaptor-TBK-binding protein 1 (TBKBP1). TBKBP1 recruits TBK1 to protein kinase C-theta (PKCθ) through a scaffold protein, CARD10. This enables PKCθ to phosphorylate TBK1 at Ser 716, a crucial step for TBK1 activation by growth factors but not by innate immune stimuli. Although the TBK1-TBKBP1 signalling axis is not required for the induction of type I IFN, it mediates mTORC1 activation and oncogenesis. Conditional deletion of either TBK1 or TBKBP1 in lung epithelial cells inhibits tumourigenesis in a mouse model of lung cancer. In addition to promoting tumour growth, the TBK1-TBKBP1 axis facilitates tumour-mediated immunosuppression through a mechanism that involves induction of the checkpoint molecule PD-L1 and stimulation of glycolysis. These findings suggest a PKCθ-TBKBP1-TBK1 growth factor signalling axis that mediates both tumour growth and immunosuppression.
- Mus musculus (House mouse),
- Immunology and Microbiology,
- Neuroscience
miR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival.
In Cell Reports on 20 August 2019 by Gagnon, J. D., Kageyama, R., et al.
PubMed
Coordinate control of T cell proliferation, survival, and differentiation are essential for host protection from pathogens and cancer. Long-lived memory cells, whose precursors are formed during the initial immunological insult, provide protection from future encounters, and their generation is the goal of many vaccination strategies. microRNAs (miRNAs) are key nodes in regulatory networks that shape effective T cell responses through the fine-tuning of thousands of genes. Here, using compound conditional mutant mice to eliminate miR-15/16 family miRNAs in T cells, we show that miR-15/16 restrict T cell cycle, survival, and memory T cell differentiation. High throughput sequencing of RNA isolated by cross-linking immunoprecipitation of AGO2 combined with gene expression analysis in miR-15/16-deficient T cells indicates that these effects are mediated through the direct inhibition of an extensive network of target genes within pathways critical to cell cycle, survival, and memory. Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
- Immunology and Microbiology,
- Mus musculus (House mouse)
High-Dimensional Characterization of IL-10 Production and IL-10-Dependent Regulation during Primary Gammaherpesvirus Infection.
In ImmunoHorizons on 21 March 2019 by Kimball, A. K., Oko, L. M., et al.
PubMed
IL-10 is a potent immunomodulatory cytokine produced by multiple cell types to restrain immune activation. Many herpesviruses use the IL-10 pathway to facilitate infection, but how endogenous IL-10 is regulated during primary infection in vivo remains poorly characterized. In this study, we infected mice with murine gammaherpesvirus 68 (γHV68) and analyzed the production and genetic contribution of IL-10 by mass cytometry (cytometry by time-of-flight) analysis. γHV68 infection elicited a breadth of effector CD4 T cells in the lungs of acutely infected mice, including a highly activated effector subset that coexpressed IFN-γ, TNF-α, and IL-10. By using IL-10 GFP transcriptional reporter mice, we identified that IL-10 was primarily expressed within CD4 T cells during acute infection in the lungs. IL10gfp-expressing CD4 T cells were highly proliferative and characterized by the expression of multiple coinhibitory receptors, including PD-1 and LAG-3. When we analyzed acute γHV68 infection of IL-10-deficient mice, we found that IL-10 limits the frequency of both myeloid and effector CD4 T cell subsets in the infected lung, with minimal changes at a distant mucosal site. These data emphasize the unique insights that high-dimensional analysis can afford in investigating antiviral immunity and provide new insights into the breadth, phenotype, and function of IL-10-expressing effector CD4 T cells during acute virus infection. Copyright © 2019 The Authors.