InVivoPlus anti-mouse IFNAR-1
Product Details
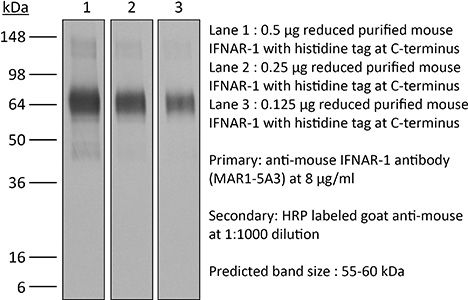
The MAR1-5A3 monoclonal antibody reacts with mouse IFNAR-1 (IFN alpha/beta receptor subunit 1). IFNAR-1 is coexpressed with IFNAR-2 on nearly all cell types and together these two subunits make up the heterodimeric Type I IFN Receptor complex. Type I IFNs (IFN-α/β) bind to the Type I IFN Receptor complex to induce cellular responses including induction of anti-viral, anti-microbial, anti-tumor, and autoimmune responses as well as to regulate the activation, proliferation, and differentiation of many cell types. The MAR1-5A3 antibody has been shown to inhibit Type I IFN receptor signaling in vitro and in vivo.Specifications
| Isotype | Mouse IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Extracellular domain of mouse IFNAR-1 |
| Reported Applications |
in vivo IFNAR-1 blockade in vitro IFNAR-1 blockade Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin* |
<1EU/mg (<0.001EU/μg) Determined by LAL gel clotting assay |
| Aggregation* |
<5% Determined by SEC |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 μM filtered |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687723 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Additional Formats

Recommended Products
-

Recommended Isotype Control(s)
InVivoPlus mouse IgG1 isotype control, unknown specificity
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo IFNAR-1 blockade
Macal, M., et al. (2018). "Self-Renewal and Toll-like Receptor Signaling Sustain Exhausted Plasmacytoid Dendritic Cells during Chronic Viral Infection" Immunity 48(4): 730-744 e735. PubMed
Although characterization of T cell exhaustion has unlocked powerful immunotherapies, the mechanisms sustaining adaptations of short-lived innate cells to chronic inflammatory settings remain unknown. During murine chronic viral infection, we found that concerted events in bone marrow and spleen mediated by type I interferon (IFN-I) and Toll-like receptor 7 (TLR7) maintained a pool of functionally exhausted plasmacytoid dendritic cells (pDCs). In the bone marrow, IFN-I compromised the number and the developmental capacity of pDC progenitors, which generated dysfunctional pDCs. Concurrently, exhausted pDCs in the periphery were maintained by self-renewal via IFN-I- and TLR7-induced proliferation of CD4(-) subsets. On the other hand, pDC functional loss was mediated by TLR7, leading to compromised IFN-I production and resistance to secondary infection. These findings unveil the mechanisms sustaining a self-perpetuating pool of functionally exhausted pDCs and provide a framework for deciphering long-term exhaustion of other short-lived innate cells during chronic inflammation.
in vivo IFNAR-1 blockade
Liu, X., et al. (2015). "CD47 blockade triggers T cell-mediated destruction of immunogenic tumors" Nat Med 21(10): 1209-1215. PubMed
Macrophage phagocytosis of tumor cells mediated by CD47-specific blocking antibodies has been proposed to be the major effector mechanism in xenograft models. Here, using syngeneic immunocompetent mouse tumor models, we reveal that the therapeutic effects of CD47 blockade depend on dendritic cell but not macrophage cross-priming of T cell responses. The therapeutic effects of anti-CD47 antibody therapy were abrogated in T cell-deficient mice. In addition, the antitumor effects of CD47 blockade required expression of the cytosolic DNA sensor STING, but neither MyD88 nor TRIF, in CD11c(+) cells, suggesting that cytosolic sensing of DNA from tumor cells is enhanced by anti-CD47 treatment, further bridging the innate and adaptive responses. Notably, the timing of administration of standard chemotherapy markedly impacted the induction of antitumor T cell responses by CD47 blockade. Together, our findings indicate that CD47 blockade drives T cell-mediated elimination of immunogenic tumors.
in vivo IFNAR-1 blockade
Yang, H., et al. (2015). "STAT3 Inhibition Enhances the Therapeutic Efficacy of Immunogenic Chemotherapy by Stimulating Type 1 Interferon Production by Cancer Cells" Cancer Res 75(18): 3812-3822. PubMed
STAT3 is an oncogenic transcription factor with potent immunosuppressive functions. We found that pharmacologic inhibition of STAT3 or its selective knockout in cancer cells improved the tumor growth-inhibitory efficacy of anthracycline-based chemotherapies. This combined effect of STAT3 inhibition/depletion and anthracyclines was only found in tumors growing on immunocompetent (not in immunodeficient) mice. As compared with Stat3-sufficient control tumors, Stat3(-/-) cancer cells exhibited an increased infiltration by dendritic cells and cytotoxic T lymphocytes after chemotherapy. Anthracyclines are known to induce several stress pathways that enhance the immunogenicity of dying and dead cancer cells, thereby stimulating a dendritic cell-dependent and T lymphocyte-mediated anticancer immune response. Among these therapy-relevant stress pathways, Stat3(-/-) cancer cells manifested one significant improvement, namely an increase in the expression of multiple type-1 interferon-responsive genes, including that of the chemokines Cxcl9 and Cxcl10. This enhanced type-1 interferon response could be suppressed by reintroducing wild-type Stat3 (but not a transactivation-deficient mutant Stat3(Y705F)) into the tumor cells. This maneuver also abolished the improved chemotherapeutic response of Stat3(-/-) cancers. Finally, the neutralization of the common type-1 interferon receptor or that of the chemokine receptor CXCR3 (which binds CXCL9 and CXCL10) abolished the difference in the chemotherapeutic response between Stat3(-/-) and control tumors. Altogether, these results suggest that STAT3 inhibitors may improve the outcome of chemotherapy by enhancing the type-1 interferon response of cancer cells.
in vitro IFNAR-1 blockade
Schliehe, C., et al. (2015). "The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection" Nat Immunol 16(1): 67-74. PubMed
Immune responses are tightly regulated to ensure efficient pathogen clearance while avoiding tissue damage. Here we report that Setdb2 was the only protein lysine methyltransferase induced during infection with influenza virus. Setdb2 expression depended on signaling via type I interferons, and Setdb2 repressed expression of the gene encoding the neutrophil attractant CXCL1 and other genes that are targets of the transcription factor NF-kappaB. This coincided with occupancy by Setdb2 at the Cxcl1 promoter, which in the absence of Setdb2 displayed diminished trimethylation of histone H3 Lys9 (H3K9me3). Mice with a hypomorphic gene-trap construct of Setdb2 exhibited increased infiltration of neutrophils during sterile lung inflammation and were less sensitive to bacterial superinfection after infection with influenza virus. This suggested that a Setdb2-mediated regulatory crosstalk between the type I interferons and NF-kappaB pathways represents an important mechanism for virus-induced susceptibility to bacterial superinfection.
in vivo IFNAR-1 blockade
Welten, S. P., et al. (2015). "The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion" Elife 4. doi : 10.7554/eLife.07486. PubMed
Signals delivered by costimulatory molecules are implicated in driving T cell expansion. The requirements for these signals, however, vary from dispensable to essential in different infections. We examined the underlying mechanisms of this differential T cell costimulation dependence and found that the viral context determined the dependence on CD28/B7-mediated costimulation for expansion of naive and memory CD8(+) T cells, indicating that the requirement for costimulatory signals is not imprinted. Notably, related to the high-level costimulatory molecule expression induced by lymphocytic choriomeningitis virus (LCMV), CD28/B7-mediated costimulation was dispensable for accumulation of LCMV-specific CD8(+) T cells because of redundancy with the costimulatory pathways induced by TNF receptor family members (i.e., CD27, OX40, and 4-1BB). Type I IFN signaling in viral-specific CD8(+) T cells is slightly redundant with costimulatory signals. These results highlight that pathogen-specific conditions differentially and uniquely dictate the utilization of costimulatory pathways allowing shaping of effector and memory antigen-specific CD8(+) T cell responses.
in vivo IFNAR-1 blockade
Ma, Y., et al. (2014). "Borrelia burgdorferi arthritis-associated locus Bbaa1 regulates Lyme arthritis and K/BxN serum transfer arthritis through intrinsic control of type I IFN production" J Immunol 193(12): 6050-6060. PubMed
Localized upregulation of type I IFN was previously implicated in development of Borrelia burgdorferi-induced arthritis in C3H mice, and was remarkable due to its absence in the mildly arthritic C57BL/6 (B6) mice. Independently, forward genetics analysis identified a quantitative trait locus on Chr4, termed B. burgdorferi-associated locus 1 (Bbaa1), that regulates Lyme arthritis severity and includes the 15 type I IFN genes. Involvement of Bbaa1 in arthritis development was confirmed in B6 mice congenic for the C3H allele of Bbaa1 (B6.C3-Bbaa1), which developed more severe Lyme arthritis and K/BxN model of rheumatoid arthritis (RA) than did parental B6 mice. Administration of a type I IFN receptor blocking mAb reduced the severity of both Lyme arthritis and RA in B6.C3-Bbaa1 mice, formally linking genetic elements within Bbaa1 to pathological production of type I IFN. Bone marrow-derived macrophages from Bbaa1 congenic mice implicated this locus as a regulator of type I IFN induction and downstream target gene expression. Bbaa1-mediated regulation of IFN-inducible genes was upstream of IFN receptor-dependent amplification; however, the overall magnitude of the response was dependent on autocrine/paracrine responses to IFN-beta. In addition, the Bbaa1 locus modulated the functional phenotype ascribed to bone marrow-derived macrophages: the B6 allele promoted expression of M2 markers, whereas the C3H allele promoted induction of M1 responses. This report identifies a genetic locus physically and functionally linked to type I IFN that contributes to the pathogenesis of both Lyme and RA.
in vivo IFNAR-1 blockade
Beug, S. T., et al. (2014). "Smac mimetics and innate immune stimuli synergize to promote tumor death" Nat Biotechnol 32(2): 182-190. PubMed
Smac mimetic compounds (SMC), a class of drugs that sensitize cells to apoptosis by counteracting the activity of inhibitor of apoptosis (IAP) proteins, have proven safe in phase 1 clinical trials in cancer patients. However, because SMCs act by enabling transduction of pro-apoptotic signals, SMC monotherapy may be efficacious only in the subset of patients whose tumors produce large quantities of death-inducing proteins such as inflammatory cytokines. Therefore, we reasoned that SMCs would synergize with agents that stimulate a potent yet safe “cytokine storm.” Here we show that oncolytic viruses and adjuvants such as poly(I:C) and CpG induce bystander death of cancer cells treated with SMCs that is mediated by interferon beta (IFN-beta), tumor necrosis factor alpha (TNF-alpha) and/or TNF-related apoptosis-inducing ligand (TRAIL). This combinatorial treatment resulted in tumor regression and extended survival in two mouse models of cancer. As these and other adjuvants have been proven safe in clinical trials, it may be worthwhile to explore their clinical efficacy in combination with SMCs.
in vivo IFNAR-1 blockade
Calame, D. G., et al. (2014). "The C5a anaphylatoxin receptor (C5aR1) protects against Listeria monocytogenes infection by inhibiting type 1 IFN expression" J Immunol 193(10): 5099-5107. PubMed
Listeria monocytogenes is a major cause of mortality resulting from food poisoning in the United States. In mice, C5 has been genetically linked to host resistance to listeriosis. Despite this genetic association, it remains poorly understood how C5 and its activation products, C5a and C5b, confer host protection to this Gram-positive intracellular bacterium. In this article, we show in a systemic infection model that the major receptor for C5a, C5aR1, is required for a normal robust host immune response against L. monocytogenes. In comparison with wild-type mice, C5aR1(-/-) mice had reduced survival and increased bacterial burden in their livers and spleens. Infected C5aR1(-/-) mice exhibited a dramatic reduction in all major subsets of splenocytes, which was associated with elevated caspase-3 activity and increased TUNEL staining. Because type 1 IFN has been reported to impede the host response to L. monocytogenes through the promotion of splenocyte death, we examined the effect of C5aR1 on type 1 IFN expression in vivo. Indeed, serum levels of IFN-alpha and IFN-beta were significantly elevated in L. monocytogenes-infected C5aR1(-/-) mice. Similarly, the expression of TRAIL, a type 1 IFN target gene and a proapoptotic factor, was elevated in NK cells isolated from infected C5aR1(-/-) mice. Treatment of C5aR1(-/-) mice with a type 1 IFNR blocking Ab resulted in near-complete rescue of L. monocytogenes-induced mortality. Thus, these findings reveal a critical role for C5aR1 in host defense against L. monocytogenes through the suppression of type 1 IFN expression.
in vivo IFNAR-1 blockade
Stock, A. T., et al. (2014). "Type I IFN suppresses Cxcr2 driven neutrophil recruitment into the sensory ganglia during viral infection" J Exp Med 211(5): 751-759. PubMed
Infection induces the expression of inflammatory chemokines that recruit immune cells to the site of inflammation. Whereas tissues such as the intestine and skin express unique chemokines during homeostasis, whether different tissues express distinct chemokine profiles during inflammation remains unclear. With this in mind, we performed a comprehensive screen of the chemokines expressed by two tissues (skin and sensory ganglia) infected with a common viral pathogen (herpes simplex virus type 1). After infection, the skin and ganglia showed marked differences in their expression of the family of Cxcr2 chemokine ligands. Specifically, Cxcl1/2/3, which in turn controlled neutrophil recruitment, was up-regulated in the skin but absent from the ganglia. Within the ganglia, Cxcl2 expression and subsequent neutrophil recruitment was inhibited by type I interferon (IFN). Using a combination of bone marrow chimeras and intracellular chemokine staining, we show that type I IFN acted by directly suppressing Cxcl2 expression by monocytes, abrogating their ability to recruit neutrophils to the ganglia. Overall, our findings describe a novel role for IFN in the direct, and selective, inhibition of Cxcr2 chemokine ligands, which results in the inhibition of neutrophil recruitment to neuronal tissue.
- Mus musculus (House mouse),
- Cancer Research,
- Immunology and Microbiology
Systemic vaccination induces CD8+ T cells and remodels the tumor microenvironment.
In Cell on 10 November 2022 by Baharom, F., Ramirez-Valdez, R. A., et al.
PubMed
Therapeutic cancer vaccines are designed to increase tumor-specific T cell immunity. However, suppressive mechanisms within the tumor microenvironment (TME) may limit T cell function. Here, we assessed how the route of vaccination alters intratumoral myeloid cells. Using a self-assembling nanoparticle vaccine that links tumor antigen peptides to a Toll-like receptor 7/8 agonist (SNP-7/8a), we treated tumor-bearing mice subcutaneously (SNP-SC) or intravenously (SNP-IV). Both routes generated antigen-specific CD8+ T cells that infiltrated tumors. However, only SNP-IV mediated tumor regression, dependent on systemic type I interferon at the time of boost. Single-cell RNA-sequencing revealed that intratumoral monocytes expressing an immunoregulatory gene signature (Chil3, Anxa2, Wfdc17) were reduced after SNP-IV boost. In humans, the Chil3+ monocyte gene signature is enriched in CD16- monocytes and associated with worse outcomes. Our results show that the generation of tumor-specific CD8+ T cells combined with remodeling of the TME is a promising approach for tumor immunotherapy. Published by Elsevier Inc.
- COVID-19,
- Immunology and Microbiology
Amphiphile-CpG vaccination induces potent lymph node activation and COVID-19 immunity in mice and non-human primates.
In NPJ Vaccines on 28 October 2022 by Seenappa, L. M., Jakubowski, A., et al.
PubMed
Despite the success of currently authorized vaccines for the reduction of severe COVID-19 disease risk, rapidly emerging viral variants continue to drive pandemic waves of infection, resulting in numerous global public health challenges. Progress will depend on future advances in prophylactic vaccine activity, including advancement of candidates capable of generating more potent induction of cross-reactive T cells and durable cross-reactive antibody responses. Here we evaluated an Amphiphile (AMP) adjuvant, AMP-CpG, admixed with SARS-CoV-2 Spike receptor binding domain (RBD) immunogen, as a lymph node-targeted protein subunit vaccine (ELI-005) in mice and non-human primates (NHPs). AMP-mediated targeting of CpG DNA to draining lymph nodes resulted in comprehensive local immune activation characterized by extensive transcriptional reprogramming, inflammatory proteomic milieu, and activation of innate immune cells as key orchestrators of antigen-directed adaptive immunity. Prime-boost immunization with AMP-CpG in mice induced potent and durable T cell responses in multiple anatomical sites critical for prophylactic efficacy and prevention of severe disease. Long-lived memory responses were rapidly expanded upon re-exposure to antigen. In parallel, RBD-specific antibodies were long-lived, and exhibited cross-reactive recognition of variant RBD. AMP-CpG-adjuvanted prime-boost immunization in NHPs was safe and well tolerated, while promoting multi-cytokine-producing circulating T cell responses cross-reactive across variants of concern (VOC). Expansion of RBD-specific germinal center (GC) B cells in lymph nodes correlated to rapid seroconversion with variant-specific neutralizing antibody responses exceeding those measured in convalescent human plasma. These results demonstrate the promise of lymph-node adjuvant-targeting to coordinate innate immunity and generate robust adaptive responses critical for vaccine efficacy. © 2022. The Author(s).
- In Vivo,
- Mus musculus (House mouse),
- Cancer Research
Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment.
In Cell on 14 October 2021 by Lam, K. C., Araya, R. E., et al.
PubMed
The tumor microenvironment (TME) influences cancer progression and therapy response. Therefore, understanding what regulates the TME immune compartment is vital. Here we show that microbiota signals program mononuclear phagocytes in the TME toward immunostimulatory monocytes and dendritic cells (DCs). Single-cell RNA sequencing revealed that absence of microbiota skews the TME toward pro-tumorigenic macrophages. Mechanistically, we show that microbiota-derived stimulator of interferon genes (STING) agonists induce type I interferon (IFN-I) production by intratumoral monocytes to regulate macrophage polarization and natural killer (NK) cell-DC crosstalk. Microbiota modulation with a high-fiber diet triggered the intratumoral IFN-I-NK cell-DC axis and improved the efficacy of immune checkpoint blockade (ICB). We validated our findings in individuals with melanoma treated with ICB and showed that the predicted intratumoral IFN-I and immune compositional differences between responder and non-responder individuals can be transferred by fecal microbiota transplantation. Our study uncovers a mechanistic link between the microbiota and the innate TME that can be harnessed to improve cancer therapies. Published by Elsevier Inc.
- Immunology and Microbiology
Knockout of MAPK Phosphatase-1 Exaggerates Type I IFN Response during Systemic Escherichia coli Infection.
In The Journal of Immunology on 15 June 2021 by Kirk, S. G., Murphy, P. R., et al.
PubMed
We have previously shown that Mkp-1-deficient mice produce elevated TNF-α, IL-6, and IL-10 following systemic Escherichia coli infection, and they exhibited increased mortality, elevated bacterial burden, and profound metabolic alterations. To understand the function of Mkp-1 during bacterial infection, we performed RNA-sequencing analysis to compare the global gene expression between E. coli-infected wild-type and Mkp-1 -/- mice. A large number of IFN-stimulated genes were more robustly expressed in E. coli-infected Mkp-1 -/- mice than in wild-type mice. Multiplex analysis of the serum cytokine levels revealed profound increases in IFN-β, IFN-γ, TNF-α, IL-1α and β, IL-6, IL-10, IL-17A, IL-27, and GMSF levels in E. coli-infected Mkp-1 -/- mice relative to wild-type mice. Administration of a neutralizing Ab against the receptor for type I IFN to Mkp-1 -/- mice prior to E. coli infection augmented mortality and disease severity. Mkp-1 -/- bone marrow-derived macrophages (BMDM) produced higher levels of IFN-β mRNA and protein than did wild-type BMDM upon treatment with LPS, E. coli, polyinosinic:polycytidylic acid, and herring sperm DNA. Augmented IFN-β induction in Mkp-1 -/- BMDM was blocked by a p38 inhibitor but not by an JNK inhibitor. Enhanced Mkp-1 expression abolished IFN-β induction by both LPS and E. coli but had little effect on the IFN-β promoter activity in LPS-stimulated RAW264.7 cells. Mkp-1 deficiency did not have an overt effect on IRF3/7 phosphorylation or IKK activation but modestly enhanced IFN-β mRNA stability in LPS-stimulated BMDM. Our results suggest that Mkp-1 regulates IFN-β production primarily through a p38-mediated mechanism and that IFN-β plays a beneficial role in E. coli-induced sepsis. Copyright © 2021 by The American Association of Immunologists, Inc.
- Mus musculus (House mouse),
- Cancer Research,
- Genetics,
- Immunology and Microbiology
DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity.
In Cancer Cell on 11 January 2021 by Lu, C., Guan, J., et al.
PubMed
Increased neoantigens in hypermutated cancers with DNA mismatch repair deficiency (dMMR) are proposed as the major contributor to the high objective response rate in anti-PD-1 therapy. However, the mechanism of drug resistance is not fully understood. Using tumor models defective in the MMR gene Mlh1 (dMLH1), we show that dMLH1 tumor cells accumulate cytosolic DNA and produce IFN-β in a cGAS-STING-dependent manner, which renders dMLH1 tumors slowly progressive and highly sensitive to checkpoint blockade. In neoantigen-fixed models, dMLH1 tumors potently induce T cell priming and lose resistance to checkpoint therapy independent of tumor mutational burden. Accordingly, loss of STING or cGAS in tumor cells decreases tumor infiltration of T cells and endows resistance to checkpoint blockade. Clinically, downregulation of cGAS/STING in human dMMR cancers correlates with poor prognosis. We conclude that DNA sensing within tumor cells is essential for dMMR-triggered anti-tumor immunity. This study provides new mechanisms and biomarkers for anti-dMMR-cancer immunotherapy. Copyright © 2020 Elsevier Inc. All rights reserved.
- Mus musculus (House mouse),
- Immunology and Microbiology
Human FcRn expression and Type I Interferon signaling control Echovirus 11 pathogenesis in mice.
In PLoS Pathogens on 1 January 2021 by Wells, A. I., Grimes, K. A., et al.
PubMed
Neonatal echovirus infections are characterized by severe hepatitis and neurological complications that can be fatal. Here, we show that expression of the human homologue of the neonatal Fc receptor (hFcRn), the primary receptor for echoviruses, and ablation of type I interferon (IFN) signaling are key host determinants involved in echovirus pathogenesis. We show that expression of hFcRn alone is insufficient to confer susceptibility to echovirus infections in mice. However, expression of hFcRn in mice deficient in type I interferon (IFN) signaling, hFcRn-IFNAR-/-, recapitulate the echovirus pathogenesis observed in humans. Luminex-based multianalyte profiling from E11 infected hFcRn-IFNAR-/- mice revealed a robust systemic immune response to infection, including the induction of type I IFNs. Furthermore, similar to the severe hepatitis observed in humans, E11 infection in hFcRn-IFNAR-/- mice caused profound liver damage. Our findings define the host factors involved in echovirus pathogenesis and establish in vivo models that recapitulate echovirus disease in humans.
- Mus musculus (House mouse),
- Cancer Research,
- Immunology and Microbiology
Autoimmunity linked protein phosphatase PTPN22 as a target for cancer immunotherapy.
In Journal for Immunotherapy of Cancer on 1 October 2020 by Cubas, R., Khan, Z., et al.
PubMed
Cancer immunotherapy has evolved from interferon-alpha (IFNα) and interleukin-2 in the 1980s to CTLA-4 and PD-1/PD-L1 checkpoint inhibitors (CPIs), the latter highlighting the importance of enhancing T-cell functions. While the search for novel immunomodulatory pathways continues, combination therapies augmenting multiple pathways can also increase efficacy. The association of autoimmune-related adverse events with clinical efficacy following CPI treatment has been inferred and suggests that breaking tolerance thresholds associated with autoimmunity may affect host immune responses for effective cancer immunotherapy. Here, we show that loss of autoimmune associated PTPN22, a key desensitization node for multiple signaling pathways, including IFNα receptor (IFNAR) and T-cell receptor, can augment tumor responses. Implantation of syngeneic tumors in Ptpn22-/- mice led to expansion and activation of peripheral and intratumoral T cells and, in turn, spontaneous tumor regression as well as enhanced responses in combination with anti-PD-L1 treatment. Using genetically modified mice expressing a catalytically inactive PTPN22 or the autoimmunity-associated human single-nucleotide polymorphism variant, augmentation of antitumor immunity was dependent on PTPN22 phosphatase activity and partially on its adaptor functions. Further, antitumor responses were dependent on both CD4+ and CD8+T cells and, in part, IFNAR function. Finally, we demonstrate that the autoimmune susceptibility Ptpn22(C1858T) variant is associated with lower risk of developing non-melanoma skin cancers, improved overall survival and increased risk for development of hyperthyroidism or hypothyroidism following atezolizumab (anti-PD-L1) treatment. Together, these data suggest that inhibition of PTPN22 phosphatase activity may provide an effective therapeutic option for cancer immunotherapy and that exploring genetic variants that shift immune tolerance thresholds may serve as a paradigm for finding new cancer immunotherapy targets. © Author(s) (or their employer(s)) 2020. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
- Immunology and Microbiology
Japanese encephalitis virus-primed CD8+ T cells prevent antibody-dependent enhancement of Zika virus pathogenesis.
In The Journal of Experimental Medicine on 7 September 2020 by Chen, D., Duan, Z., et al.
PubMed
Cross-reactive anti-flaviviral immunity can influence the outcome of infections with heterologous flaviviruses. However, it is unclear how the interplay between cross-reactive antibodies and T cells tilts the balance toward pathogenesis versus protection during secondary Zika virus (ZIKV) and Japanese encephalitis virus (JEV) infections. We show that sera and IgG from JEV-vaccinated humans and JEV-inoculated mice cross-reacted with ZIKV, exacerbated lethal ZIKV infection upon transfer to mice, and promoted viral replication and mortality upon ZIKV infection of the neonates born to immune mothers. In contrast, transfer of CD8+ T cells from JEV-exposed mice was protective, reducing the viral burden and mortality of ZIKV-infected mice and abrogating the lethal effects of antibody-mediated enhancement of ZIKV infection in mice. Conversely, cross-reactive anti-ZIKV antibodies or CD8+ T cells displayed the same pathogenic or protective effects upon JEV infection, with the exception that maternally acquired anti-ZIKV antibodies had no effect on JEV infection of the neonates. These results provide clues for developing safe anti-JEV/ZIKV vaccines. © 2020 Ningbo University.
- Cancer Research,
- Immunology and Microbiology
Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP.
In Immunity on 18 February 2020 by Zhou, Y., Fei, M., et al.
PubMed
Clearance of apoptotic cells by macrophages prevents excessive inflammation and supports immune tolerance. Here, we examined the effect of blocking apoptotic cell clearance on anti-tumor immune response. We generated an antibody that selectively inhibited efferocytosis by phagocytic receptor MerTK. Blockade of MerTK resulted in accumulation of apoptotic cells within tumors and triggered a type I interferon response. Treatment of tumor-bearing mice with anti-MerTK antibody stimulated T cell activation and synergized with anti-PD-1 or anti-PD-L1 therapy. The anti-tumor effect induced by anti-MerTK treatment was lost in Stinggt/gt mice, but not in Cgas-/- mice. Abolishing cGAMP production in Cgas-/- tumor cells, depletion of extracellular ATP, or inactivation of the ATP-gated P2X7R channel also compromised the effects of MerTK blockade. Mechanistically, extracellular ATP acted via P2X7R to enhance the transport of extracellular cGAMP into macrophages and subsequent STING activation. Thus, MerTK blockade increases tumor immunogenicity and potentiates anti-tumor immunity, which has implications for cancer immunotherapy. Copyright © 2020 Elsevier Inc. All rights reserved.
- Immunology and Microbiology
Systemic inflammation suppresses lymphoid tissue remodeling and B cell immunity during concomitant local infection
Preprint on BioRxiv : the Preprint Server for Biology on 6 November 2019 by Alexandre, Y. O., Devi, S., et al.
PubMed
Concurrent infection with multiple pathogens occurs frequently in individuals and can result in exacerbated infections and altered immunity. However, the impact of such coinfections on immune responses remains poorly understood. Here we reveal that systemic infection results in an inflammation-induced suppression of local immunity. During localized infection or vaccination in barrier tissues including the skin or respiratory tract, concurrent systemic infection induced a type I interferon-dependent lymphopenia that impairs lymphocyte recruitment to the draining lymph node (dLN). This leads to suppressed lymphoid stromal cell expansion and dLN remodeling and impaired induction of B cell responses and antibody production. Our data suggest that contemporaneous systemic inflammation constrains the induction of regional immunity.
Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice.
In Proceedings of the National Academy of Sciences of the United States of America on 5 February 2019 by Wang, A., Pope, S. D., et al.
PubMed
Secondary hemophagocytic lymphohistiocytosis (sHLH) is a highly mortal complication associated with sepsis. In adults, it is often seen in the setting of infections, especially viral infections, but the mechanisms that underlie pathogenesis are unknown. sHLH is characterized by a hyperinflammatory state and the presence hemophagocytosis. We found that sequential challenging of mice with a nonlethal dose of viral toll-like receptor (TLR) agonist followed by a nonlethal dose of TLR4 agonist, but not other permutations, produced a highly lethal state that recapitulates many aspects of human HLH. We found that this hyperinflammatory response could be recapitulated in vitro in bone marrow-derived macrophages. RNA sequencing analyses revealed dramatic up-regulation of the red-pulp macrophage lineage-defining transcription factor SpiC and its associated transcriptional program, which was also present in bone marrow macrophages sorted from patients with sHLH. Transcriptional profiling also revealed a unique metabolic transcriptional profile in these macrophages, and immunometabolic phenotyping revealed impaired mitochondrial function and oxidative metabolism and a reliance on glycolytic metabolism. Subsequently, we show that therapeutic administration of the glycolysis inhibitor 2-deoxyglucose was sufficient to rescue animals from HLH. Together, these data identify a potential mechanism for the pathogenesis of sHLH and a potentially useful therapeutic strategy for its treatment.
- Mus musculus (House mouse),
- Immunology and Microbiology
Alcohol enhances type 1 interferon-α production and mortality in young mice infected with Mycobacterium tuberculosis.
In PLoS Pathogens on 1 August 2018 by Tripathi, D., Welch, E., et al.
PubMed
In the current study, we used a mouse model and human blood samples to determine the effects of chronic alcohol consumption on immune responses during Mycobacterium tuberculosis (Mtb) infection. Alcohol increased the mortality of young mice but not old mice with Mtb infection. CD11b+Ly6G+ cells are the major source of IFN-α in the lungs of Mtb-infected alcohol-fed young mice, and IFN-α enhances macrophage necroptosis in the lungs. Treatment with an anti-IFNAR-1 antibody enhanced the survival of Mtb-infected alcohol-fed young mice. In response to Mtb, peripheral blood mononuclear cells (PBMCs) from alcoholic young healthy individuals with latent tuberculosis infection (LTBI) produced significantly higher amounts of IFN-α than those from non-alcoholic young healthy LTBI+ individuals and alcoholic and non-alcoholic old healthy LTBI+ individuals. Our study demonstrates that alcohol enhances IFN-α production by CD11b+Ly6G+ cells in the lungs of young Mtb-infected mice, which leads to macrophage necroptosis and increased mortality. Our findings also suggest that young alcoholic LTBI+ individuals have a higher risk of developing active TB infection.
- Cancer Research
TGFβ blocks IFNα/β release and tumor rejection in spontaneous mammary tumors
Preprint on BioRxiv : the Preprint Server for Biology on 1 June 2018 by Guerin, M. V., Regnier, F., et al.
PubMed
h4>Summary/h4> Type I interferons (IFN) are being rediscovered as potent anti-tumoral agents. Activation of the STimulator of INterferon Genes (STING) by DMXAA can induce a strong production of IFNα/β and the rejection of transplanted primary tumors. In the present study, we addressed whether targeting STING with DMXAA also leads to the regression of spontaneous MMTV-PyMT mammary tumors. We show that these tumors are refractory to DMXAA-induced regression. This is due to a blockade in the phosphorylation of IRF3 and the ensuing IFNα/β production. Mechanistically, we identified TGFβ abundant in spontaneous tumors, as a key molecule limiting this IFN-induced-tumor regression by DMXAA. Finally, blocking TGFβ restores the production of IFNα by activated MHCII + tumor-associated macrophages, and enables tumor regression induced by STING activation. On the basis of these findings, we propose that type I IFN-dependent cancer therapies could be greatly improved by combinations including the blockade of TGFβ.