InVivoMAb anti-mouse/rat IL-1β
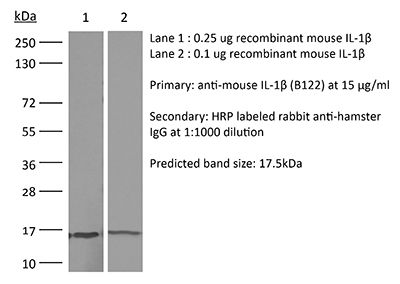
Product Description
Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse IL-1β |
| Reported Applications |
in vivo IL-1β neutralization in vitro IL-1β neutralization ELISA |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687727 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
-
Chen, K. W., et al (2014). "The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge" Cell Rep 8(2): 570-582.
PubMed
The macrophage NLRC4 inflammasome drives potent innate immune responses against Salmonella by eliciting caspase-1-dependent proinflammatory cytokine production (e.g., interleukin-1beta [IL-1beta]) and pyroptotic cell death. However, the potential contribution of other cell types to inflammasome-mediated host defense against Salmonella was unclear. Here, we demonstrate that neutrophils, typically viewed as cellular targets of IL-1beta, themselves activate the NLRC4 inflammasome during acute Salmonella infection and are a major cell compartment for IL-1beta production during acute peritoneal challenge in vivo. Importantly, unlike macrophages, neutrophils do not undergo pyroptosis upon NLRC4 inflammasome activation. The resistance of neutrophils to pyroptotic death is unique among inflammasome-signaling cells so far described and allows neutrophils to sustain IL-1beta production at a site of infection without compromising the crucial inflammasome-independent antimicrobial effector functions that would be lost if neutrophils rapidly lysed upon caspase-1 activation. Inflammasome pathway modification in neutrophils thus maximizes host proinflammatory and antimicrobial responses during pathogen challenge.
-
Gopinath, S., et al (2014). "Role of disease-associated tolerance in infectious superspreaders" Proc Natl Acad Sci U S A 111(44): 15780-15785.
PubMed
Natural populations show striking heterogeneity in their ability to transmit disease. For example, a minority of infected individuals known as superspreaders carries out the majority of pathogen transmission events. In a mouse model of Salmonella infection, a subset of infected hosts becomes superspreaders, shedding high levels of bacteria (>10(8) cfu per g of feces) but remain asymptomatic with a dampened systemic immune state. Here we show that superspreader hosts remain asymptomatic when they are treated with oral antibiotics. In contrast, nonsuperspreader Salmonella-infected hosts that are treated with oral antibiotics rapidly shed superspreader levels of the pathogen but display signs of morbidity. This morbidity is linked to an increase in inflammatory myeloid cells in the spleen followed by increased production of acute-phase proteins and proinflammatory cytokines. The degree of colonic inflammation is similar in antibiotic-treated superspreader and nonsuperspreader hosts, indicating that the superspreader hosts are tolerant of antibiotic-mediated perturbations in the intestinal tract. Importantly, neutralization of acute-phase proinflammatory cytokines in antibiotic-induced superspreaders suppresses the expansion of inflammatory myeloid cells and reduces morbidity. We describe a unique disease-associated tolerance to oral antibiotics in superspreaders that facilitates continued transmission of the pathogen.
Product Citations
-
TREM2 sustains glucose metabolic homeostasis to drive antibacterial defense during sepsis.
In iScience on 17 April 2026 by Wu, Z., Wang, X., et al.
PubMed
Metabolic disturbances, particularly glucose imbalances, are common in sepsis and are strongly associated with increased mortality. However, the mechanisms underlying glucose dyshomeostasis remain poorly understood. Here, we revealed the role of triggering receptor expressed on myeloid cells 2 (TREM2) in regulating glucose metabolism during sepsis. Macrophage-specific TREM2 deficiency significantly increased the level of abdominal IL-1β, which is predominantly released by pyroptotic peritoneal macrophages. IL-1β then acts on IL-1R1 receptors on pancreatic islet β-cells, promoting insulin release and inducing hypoglycemia. Transfusing TREM2-overexpressing macrophages and administering glucose solutions can restore glucose homeostasis and improve sepsis outcomes in mice. In summary, our study reveals a mechanism by which TREM2 orchestrates glucose metabolism during sepsis and highlights the potential of TREM2 as a therapeutic target for sepsis.
-
GSDME-IL-18 pyroptotic axis prevents myosteatosis by expanding tissue-resident macrophages to promote muscle regeneration.
In J Clin Invest on 15 April 2026 by Cao, Q., Liu, J., et al.
PubMed
Metabolic-inflammatory crosstalk orchestrates muscle repair. Although pyroptosis typically aggravates sterile injury, we demonstrated that GSDME-dependent pyroptotic signaling associated with recruited myeloid cells paradoxically supported regeneration. GSDME expression was induced in postsurgical human muscle injury and murine damage models. Gsdme deficiency delayed functional recovery and exacerbated injury-induced myosteatosis, a pathological form of intramuscular ectopic fat deposition. Time-series and scRNA-seq analyses revealed that GSDME loss shifted the transcriptional program from oxidative metabolism to lipid storage and adipogenesis. Lipidomics confirmed aberrant accumulation of triacylglycerols (TAGs) and sphingolipids in Gsdme-deficient muscle. Single-cell profiling further identified divergent fibro-adipogenic progenitor (FAP) states skewed toward adipogenesis, accompanied by impaired expansion of restorative Lyve1+Cd163+Txnip+ tissue-resident macrophages (TRMs), as validated by multiplex flow cytometry. Blocking CCR2-dependent monocyte recruitment produced regenerative defects comparable with those caused by Gsdme deficiency. Myeloid-specific Gsdme reintroduction rescued TRM expansion and function and curbed FAP adipogenic reprogramming, whereas FAP-specific expression proved ineffective. Mechanistically, IL-18 downstream of GSDME-dependent signaling engaged KLF4/JUN signaling in TRMs, sustaining their reparative and lipid-clearing capacity. This GSDME-IL-18-TRM axis was compromised in aged muscle, yet exogenous IL-18 reversed myosteatosis and accelerated regeneration. Together, these findings suggest that GSDME-dependent pyroptotic signaling can act as a metabolic checkpoint that sustains TRM-driven lipid homeostasis to support muscle regeneration.
-
Targeting CD177: A Novel Therapeutic Strategy for NLRP3-Associated Autoinflammatory Diseases.
In Int J Mol Sci on 20 March 2026 by Zhu, Y., Zhang, F., et al.
PubMed
NLRP3-associated autoinflammatory diseases (NLRP3-AIDs) are rare autoinflammatory disorders caused by uncontrolled inflammasome activation. While IL-1β blockade is first-line therapy, many patients respond inadequately, highlighting a need for alternative strategies. Transcriptomic analysis was performed on immune cells from a patient with an NLRP3 L573W mutation. Functional validation of CD177 as a downstream effector of NLRP3 activation was conducted. A novel NLRP3 L573W knock-in mouse model was established. Correlation between CD177 expression, disease severity, neutrophilia, and tissue damage was assessed. Therapeutic efficacy of siRNA-mediated CD177 silencing was evaluated and compared with IL-1β blockade. CD177, a neutrophil-specific protein, was significantly upregulated in NLRP3-mutant cells and confirmed as a direct downstream effector of NLRP3 activation. The NLRP3 L573W knock-in mouse recapitulated human disease heterogeneity, from mild self-limited inflammation to severe multi-organ pathology. CD177 expression correlated with disease severity, neutrophilia, and tissue damage. siRNA-mediated CD177 silencing attenuated systemic inflammation, reduced neutrophil infiltration and cytokine levels (IL-1β, IL-6, TNFα), and ameliorated multi-organ damage, with effects comparable to or exceeding those of IL-1β blockade. CD177 is a non-canonical amplifier of NLRP3-driven inflammation. Targeting CD177 represents a superior therapeutic strategy for NLRP3-AIDs, including IL-1β-refractory cases.
-
Microglial NLRP3-dependent pyroptosis promotes cognitive dysfunction of diabetic encephalopathy by inhibiting adult hippocampal neurogenesis through the release of IL-1β.
In Acta Pharmacol Sin on 20 March 2026 by Hua, M. Y., Huang, S., et al.
PubMed
Diabetic encephalopathy (DE) is a prevalent complication of diabetes which can lead to cognitive dysfunction, without effective therapy currently. In diabetic patients, a reduction in adult hippocampal neurogenesis (AHN) is a heightened risk of cognitive impairment, which may be associated with neuroinflammation caused by microglia. In this study, we established a DE mouse model and conducted in vitro cultures of microglial cells and neural stem cells. Our study demonstrated that the high-glucose associated with DE impairs AHN and induces microglial NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) dependent pyroptosis. Further investigation showed that upregulation of microglial NLRP3 promotes the activation of Gasdermin D (GSDMD), the key pyroptosis effector, and the cleavage of pro-interleukin-1β (pro-IL-1β) by caspase-1, exacerbated pyroptosis and induced release of IL-1β, which might lead to impaired AHN and subsequent cognitive dysfunction. Conversely, downregulation of microglial NLRP3 inhibited caspase-1 activation and pyroptosis, reduced release of IL-1β, improved AHN, and rescued cognitive deficits in DE mouse model. Such findings suggest that targeting microglial NLRP3 inflammasome-mediated pyroptosis may be an important potential therapeutic target for treating DE.