InVivoMAb anti-mouse F4/80
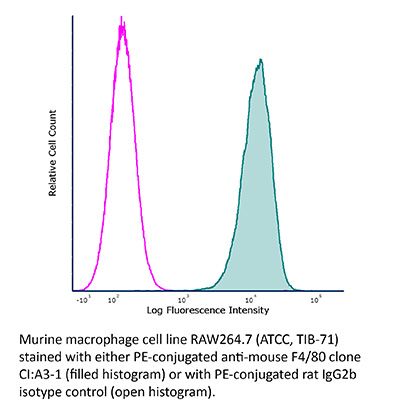
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | C57BL/6 mouse thioglycollate stimulated peritoneal macrophages |
| Reported Applications |
in vivo Monocyte/Macrophage depletion Functional assays Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10949019 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Wang, W., et al (2018). "RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer" Cancer Cell 34(5): 757-774 e757.
PubMed
Pancreatic ductal adenocarcinoma (PDA) is characterized by immune tolerance and immunotherapeutic resistance. We discovered upregulation of receptor-interacting serine/threonine protein kinase 1 (RIP1) in tumor-associated macrophages (TAMs) in PDA. To study its role in oncogenic progression, we developed a selective small-molecule RIP1 inhibitor with high in vivo exposure. Targeting RIP1 reprogrammed TAMs toward an MHCII(hi)TNFalpha(+)IFNgamma(+) immunogenic phenotype in a STAT1-dependent manner. RIP1 inhibition in TAMs resulted in cytotoxic T cell activation and T helper cell differentiation toward a mixed Th1/Th17 phenotype, leading to tumor immunity in mice and in organotypic models of human PDA. Targeting RIP1 synergized with PD1-and inducible co-stimulator-based immunotherapies. Tumor-promoting effects of RIP1 were independent of its co-association with RIP3. Collectively, our work describes RIP1 as a checkpoint kinase governing tumor immunity.
-
Paul, S., et al (2019). "Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor" J Immunother Cancer 7(1): 208.
PubMed
BACKGROUND: NKT cells play an important role in anti-tumor immunity. Alpha-galactosylceramide (α-GalCer), a synthetic glycolipid is presented to natural killer T (NKT) cells by most antigen-presenting cells through CD1d molecules leading to activation of NKT cells. However, the precise mechanisms of how α-GalCer-activated NKT regulate the polarization of the macrophages and effector T cells in the solid tumor are not studied adequately. METHODS: We induced solid tumor in C57BL/6 mice by subcutaneous injection of B16F10 cell line (1 X 10(6) cells) and monitored the tumor growth. Animals were given an intraperitoneal injection of α-GalCer (2 μg/injection) in 200 μl PBS on day + 1, + 5, + 10, + 15, and + 20 (with respect to tumor cell injection). Immune cells were characterized using flow cytometry and immunofluorescence staining. NK cells, Gr1(+) cells, and F4/80(+) macrophages in the mice were depleted by intravenous injection of cell-specific antibodies. Statistical analysis was performed using Student’s t-test or one-way ANOVA. RESULTS: Our results showed that intratumoral NKT cells have a lower frequency of CD69, CD25, CD122, and IFN-γR expression; produced less inflammatory cytokines such as IFN-γ, TNF-α, and GM-CSF; higher frequency CD62L(+) NKT cells; and also showed reduced proliferation as compared to the splenic NKT cells. Mice treated with α-GalCer showed a significantly increased frequency of IFN-γ-producing NKT cells, CD8(+) T cells, and effector Th1 cells. Depletion of NK cells in α-GalCer-treated mice showed a lower frequency of IFN-γ-producing CD4(+) and CD8(+) T cells in the tumor and prevented the α-GalCer-induced tumor growth. NKT cell activation with α-GalCer treatment significantly increased the iNOS(+)CD206(-) M1-macrophages and reduced the iNOS(-)CD206(+) M2-macrophages in the spleen and tumor, and depletion of F4/80(+) macrophages prevented the α-GalCer-induced reduction in the tumor growth. CONCLUSIONS: We showed that activation of NKT cell with α-GalCer modulates the frequency of M1-macrophages and effector Th1 cells in the secondary lymphoid tissues and tumor microenvironment and inhibit tumor growth. The finding suggests that activation of NKT cells with α-GalCer may provide an effective anti-cancer outcome.
-
Wang, E. C. E., et al (2019). "A Subset of TREM2(+) Dermal Macrophages Secretes Oncostatin M to Maintain Hair Follicle Stem Cell Quiescence and Inhibit Hair Growth" Cell Stem Cell 24(4): 654-669.e656.
PubMed
Hair growth can be induced from resting mouse hair follicles by topical application of JAK inhibitors, suggesting that JAK-STAT signaling is required for maintaining hair follicle stem cells (HFSCs) in a quiescent state. Here, we show that Oncostatin M (OSM), an IL-6 family cytokine, negatively regulates hair growth by signaling through JAK-STAT5 to maintain HFSC quiescence. Genetic deletion of the OSM receptor or STAT5 can induce premature HFSC activation, suggesting that the resting telogen stage is actively maintained by the hair follicle niche. Single-cell RNA sequencing revealed that the OSM source is not intrinsic to the hair follicle itself and is instead a subset of TREM2(+) macrophages that is enriched within the resting follicle and deceases immediately prior to HFSC activation. In vivo inhibition of macrophage function was sufficient to induce HFSC proliferation and hair cycle induction. Together these results clarify how JAK-STAT signaling actively inhibits hair growth.
-
Albacker, L. A., et al (2013). "TIM-4, expressed by medullary macrophages, regulates respiratory tolerance by mediating phagocytosis of antigen-specific T cells" Mucosal Immunol 6(3): 580-590.
PubMed
Respiratory exposure to antigen induces T cell tolerance via several overlapping mechanisms that limit the immune response. While the mechanisms involved in the development of Treg cells have received much attention, those that result in T cell deletion are largely unknown. Herein, we show that F4/80(+) lymph node medullary macrophages expressing TIM-4, a phosphatidylserine receptor, remove antigen-specific T cells during respiratory tolerance, thereby reducing secondary T cell responses. Blockade of TIM-4 inhibited the phagocytosis of antigen-specific T cells by TIM-4 expressing lymph node medullary macrophages, resulting in an increase in the number of antigen-specific T cells and the abrogation of respiratory tolerance. Moreover, specific depletion of medullary macrophages inhibited the induction of respiratory tolerance, highlighting the key role of TIM-4 and medullary macrophages in tolerance. Therefore, TIM-4-mediated clearance of antigen specific T cells represents an important previously unrecognized mechanism regulating respiratory tolerance.
Product Citations
-
Natural killer T cell activation increases iNOS+CD206- M1 macrophage and controls the growth of solid tumor.
In Journal for Immunotherapy of Cancer on 6 August 2019 by Paul, S., Chhatar, S., et al.
PubMed
NKT cells play an important role in anti-tumor immunity. Alpha-galactosylceramide (α-GalCer), a synthetic glycolipid is presented to natural killer T (NKT) cells by most antigen-presenting cells through CD1d molecules leading to activation of NKT cells. However, the precise mechanisms of how α-GalCer-activated NKT regulate the polarization of the macrophages and effector T cells in the solid tumor are not studied adequately. We induced solid tumor in C57BL/6 mice by subcutaneous injection of B16F10 cell line (1 X 106 cells) and monitored the tumor growth. Animals were given an intraperitoneal injection of α-GalCer (2 μg/injection) in 200 μl PBS on day + 1, + 5, + 10, + 15, and + 20 (with respect to tumor cell injection). Immune cells were characterized using flow cytometry and immunofluorescence staining. NK cells, Gr1+ cells, and F4/80+ macrophages in the mice were depleted by intravenous injection of cell-specific antibodies. Statistical analysis was performed using Student's t-test or one-way ANOVA. Our results showed that intratumoral NKT cells have a lower frequency of CD69, CD25, CD122, and IFN-γR expression; produced less inflammatory cytokines such as IFN-γ, TNF-α, and GM-CSF; higher frequency CD62L+ NKT cells; and also showed reduced proliferation as compared to the splenic NKT cells. Mice treated with α-GalCer showed a significantly increased frequency of IFN-γ-producing NKT cells, CD8+ T cells, and effector Th1 cells. Depletion of NK cells in α-GalCer-treated mice showed a lower frequency of IFN-γ-producing CD4+ and CD8+ T cells in the tumor and prevented the α-GalCer-induced tumor growth. NKT cell activation with α-GalCer treatment significantly increased the iNOS+CD206- M1-macrophages and reduced the iNOS-CD206+ M2-macrophages in the spleen and tumor, and depletion of F4/80+ macrophages prevented the α-GalCer-induced reduction in the tumor growth. We showed that activation of NKT cell with α-GalCer modulates the frequency of M1-macrophages and effector Th1 cells in the secondary lymphoid tissues and tumor microenvironment and inhibit tumor growth. The finding suggests that activation of NKT cells with α-GalCer may provide an effective anti-cancer outcome.
-
The Ly6ghigh Neutrophil Subset Dictates Breast Cancer Lung Metastasis via CD8+ T Cell Death.
In Cancer Commun (Lond) on 2 February 2026 by Wang, R., Liu, X., et al.
PubMed
Background: Lung metastasis is a leading cause of breast cancer (BC)-related mortality, driven by the immunosuppressive traits of the metastatic tumor microenvironment. However, the mechanisms underlying cell-cell crosstalk in shaping immune evasion within the metastatic niche remain poorly defined. Neutrophil extracellular traps (NETs) and their associated proteins, such as cathelicidin, have emerged as key mediators of metastatic regulation in cancer. Here, we aimed to decipher the interaction between a neutrophil subset characterized by high expression of lymphocyte antigen 6 complex locus g (Ly6ghigh) and cluster of differentiation 8-positive T lymphocytes (CD8+ T cells), mediated via cathelicidin embedded in NETs, as well as their synergistic mechanism and cooperative role in promoting lung metastasis of BC. Methods: We characterized neutrophil heterogeneity and functional dynamics by performing single-cell RNA sequencing and flow cytometry on lung tissues derived from murine models of BC lung metastasis. We utilized cathelicidin-related antimicrobial peptide (Cramp) knockout mice to dissect the role of cathelicidin in NETs. The spatial colocalization of apoptotic CD8+ T cells and NETs was analyzed using multiplex immunofluorescence, and the molecular interactions were probed by protein binding assays. Results: Neutrophils in the lung metastatic niche were classified into 2 subsets based on the Ly6g expression: Ly6ghigh and Ly6glow neutrophils. Ly6glow neutrophils, which were recruited in the macrometastatic stage, exhibited myeloid-derived suppressor cell-like characteristics. Notably, Ly6ghigh neutrophils induced CD8+ T cell apoptosis through NET formation, with apoptotic CD8+ T cells spatially clustered within NET-rich areas. Mechanistically, NET-derived cathelicidin (Cramp in mice) directly bound to mitochondrial adenine nucleotide translocator 1 (Ant1) in CD8+ T cells, triggering conformational changes and complex formation with voltage-dependent anion channel 1 (Vdac1). These events resulted in the opening of the mitochondrial permeability transition pore and loss of mitochondrial membrane potential. Conclusions: Our study demonstrates that Ly6ghigh neutrophils play a critical role in immunosuppression and immune evasion through NET-induced apoptosis of CD8+ T cells. These findings underscore the importance of NETs and cathelicidin in BC lung metastasis, suggesting their potential as therapeutic targets in restoring antitumor immunity and in preventing metastatic progression.
-
FLT3L combined with GM-CSF induced dendritic cells drive broad tumor-specific CD8+ T cell responses and remodel the tumor microenvironment to enhance anti-tumor efficacy.
In Front Immunol on 22 September 2025 by Zheng, Q., Zhang, J., et al.
PubMed
Dendritic cells (DCs) play a crucial role in anti-tumor immunity by capturing, processing, and presenting tumor antigens to T cells, making DC-based immunotherapy a promising approach for cancer treatment. However, the most commonly used clinical strategy still relies on inducing DCs in vitro using granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL - 4) (GM/IL4-DCs), which often results in a heterogeneous cell population with suboptimal anti-tumor function. Here, we compared DCs generated by co-stimulating with FMS-like tyrosine kinase 3 ligand (FLT3L) and GM-CSF (FL/GM-DCs) with the conventional GM/IL4-DCs.
-
Toll-like receptor 7 (TLR7)-mediated antiviral response protects mice from lethal SARS-CoV-2 infection.
In J Virol on 20 May 2025 by Ghimire, R., Shrestha, R., et al.
PubMed
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced impaired antiviral immunity and excessive inflammatory responses cause lethal pneumonia. However, the in vivo roles of key pattern recognition receptors that elicit protective antiviral and fatal inflammatory responses, specifically in the lungs, are not well described. Coronaviruses possess single-stranded RNA genome that activates TLR7/8 to induce an antiviral interferon (IFN) and robust inflammatory cytokine response. Here, using wild-type and TLR7-deficient (TLR7-/-) mice infected with mouse-adapted SARS-CoV-2 (MA-CoV-2), we examined the role of TLR7 in the lung antiviral and inflammatory response and severe pneumonia. We showed that TLR7 deficiency significantly increased lung virus loads and morbidity/mortality, which correlated with reduced levels of type I IFNs (Ifna/b), type III IFNs (Ifnl), and IFN-stimulated genes (ISGs) in the lungs. A detailed evaluation of MA-CoV-2-infected lungs revealed increased neutrophil accumulation and lung pathology in TLR7-/- mice. We further showed that blocking type I IFN receptor (IFNAR) signaling enhanced SARS-CoV-2 replication in the lungs and caused severe lung pathology, leading to 100% mortality compared to infected control mice. Moreover, immunohistochemical assessment of the lungs revealed increased numbers of SARS-CoV-2 antigen-positive macrophages, pneumocytes, and bronchial epithelial cells in TLR7-/- and IFNAR-deficient mice compared to control mice. In summary, we conclusively demonstrated that despite TLR7-induced robust lung inflammation, TLR7-induced IFN/ISG responses suppress lung virus replication and pathology and provide protection against SARS-CoV-2-induced fatal pneumonia. Additionally, given the similar disease outcomes in control, TLR7-/-, and IFNAR-deficient MA-CoV-2-infected mice and coronavirus disease 2019 (COVID-19) patients, we propose that MA-CoV-2-infected mice constitute an excellent model for studying COVID-19.IMPORTANCESevere coronavirus disease 2019 (COVID-19) is caused by a delicate balance between a strong antiviral and an exuberant inflammatory response. A robust antiviral immunity and regulated inflammation are protective, while a weak antiviral response and excessive inflammation are detrimental. However, the key host immune sensors that elicit protective antiviral and inflammatory responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) challenge are poorly defined. Here, we examined the role of viral RNA-mediated TLR7 activation in the lung antiviral and inflammatory responses in SARS-CoV-2-infected mice. We demonstrate that TLR7 deficiency led to a high rate of morbidity and mortality, which correlated with an impaired antiviral interferon (IFN)-I/III response, enhanced lung virus replication, and severe lung pathology. Furthermore, we show that blocking IFN-I signaling using anti-IFN receptor antibody promoted SARS-CoV-2 replication in the lungs and caused severe disease. These results provide conclusive evidence that TLR7 and IFN-I receptor deficiencies lead to severe disease in mice, replicating clinical features observed in COVID-19 patients.