InVivoMAb anti-human CD11a (LFA-1α)
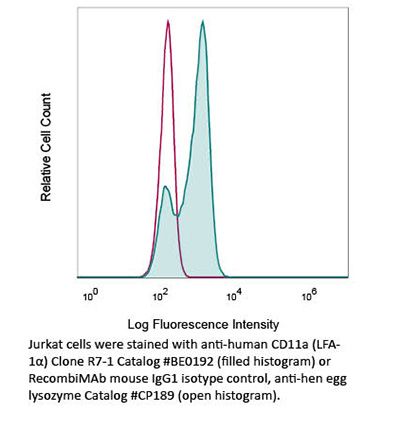
Product Description
Specifications
| Isotype | Mouse IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb mouse IgG1 isotype control, unknown specificity |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications |
Functional assays Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10948991 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Welzenbach, K., et al (2015). "A novel multi-parameter assay to dissect the pharmacological effects of different modes of integrin alphaLbeta2 inhibition in whole blood" Br J Pharmacol 172(20): 4875-4887.
PubMed
BACKGROUND AND PURPOSE: The integrin alphaLbeta2 plays central roles in leukocyte adhesion and T cell activation, rendering alphaLbeta2 an attractive therapeutic target. Compounds with different modes of alphaLbeta2 inhibition are in development, currently. Consequently, there is a foreseeable need for bedside assays, which allow assessment of the different effects of diverse types of alphaLbeta2 inhibitors in the peripheral blood of treated patients. EXPERIMENTAL APPROACH: Here, we describe a flow cytometry-based technology that simultaneously quantitates alphaLbeta2 conformational change upon inhibitor binding, alphaLbeta2 expression and T cell activation at the single-cell level in human blood. Two classes of allosteric low MW inhibitors, designated alpha I and alpha/beta I allosteric alphaLbeta2 inhibitors, were investigated. The first application revealed intriguing inhibitor class-specific profiles. KEY RESULTS: Half-maximal inhibition of T cell activation was associated with 80% epitope loss induced by alpha I allosteric inhibitors and with 40% epitope gain induced by alpha/beta I allosteric inhibitors. This differential establishes that inhibitor-induced alphaLbeta2 epitope changes do not directly predict the effect on T cell activation. Moreover, we show here for the first time that alpha/beta I allosteric inhibitors, in contrast to alpha I allosteric inhibitors, provoked partial downmodulation of alphaLbeta2, revealing a novel property of this inhibitor class. CONCLUSIONS AND IMPLICATIONS: The multi-parameter whole blood alphaLbeta2 assay described here may enable therapeutic monitoring of alphaLbeta2 inhibitors in patients’ blood. The assay dissects differential effect profiles of different classes of alphaLbeta2 inhibitors.
-
Stark, K., et al (2013). "Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs" Nat Immunol 14(1): 41-51.
PubMed
Coordinated navigation within tissues is essential for cells of the innate immune system to reach the sites of inflammatory processes, but the signals involved are incompletely understood. Here we demonstrate that NG2(+) pericytes controlled the pattern and efficacy of the interstitial migration of leukocytes in vivo. In response to inflammatory mediators, pericytes upregulated expression of the adhesion molecule ICAM-1 and released the chemoattractant MIF. Arteriolar and capillary pericytes attracted and interacted with myeloid leukocytes after extravasating from postcapillary venules, ‘instructing’ them with pattern-recognition and motility programs. Inhibition of MIF neutralized the migratory cues provided to myeloid leukocytes by NG2(+) pericytes. Hence, our results identify a previously unknown role for NG2(+) pericytes as an active component of innate immune responses, which supports the immunosurveillance and effector function of extravasated neutrophils and macrophages.
-
Welzenbach, K., et al (2002). "Small molecule inhibitors induce conformational changes in the I domain and the I-like domain of lymphocyte function-associated antigen-1. Molecular insights into integrin inhibition" J Biol Chem 277(12): 10590-10598.
PubMed
The beta(2) integrin lymphocyte function-associated antigen-1 (LFA-1) is a conformationally flexible alpha/beta heterodimeric receptor, which is expressed on the surface of all leukocytes. LFA-1 mediates cell adhesion crucial for normal immune and inflammatory responses. Intracellular signals or cations are required to convert LFA-1 from a nonligand binding to a ligand binding state. Here we investigated the effect of small molecule inhibitors on LFA-1 by monitoring the binding of monoclonal antibodies mapped to different receptor domains. The inhibitors were found to not only induce epitope changes in the I domain of the alpha(L) chain but also in the I-like domain of the beta(2) chain depending on the individual chemical structure of the inhibitor and its binding site. For the first time, we provide strong evidence that the I-like domain represents a target for allosteric LFA-1 inhibition similar to the well established regulatory L-site on the I domain of LFA-1. Moreover, the antibody binding patterns observed in the presence of the various inhibitors establish a conformational interaction between the LFA-1 I domain and the I-like domain in the native receptor that is formed upon activation. Differentially targeting the binding sites of the inhibitors, the L-site and the I-like domain, may open new avenues for highly specific therapeutic intervention in diseases where integrins play a pathophysiological role.
-
Lu, H., et al (2000). "LFA-1 (CD11a/CD18) triggers hydrogen peroxide production by canine neutrophils" J Leukoc Biol 68(1): 73-80.
PubMed
The respiratory burst of neutrophils stimulated by chemotactic factors is markedly augmented by Mac-1-dependent adhesion such as the interaction of Mac-1 (CD11b/CD18) with intercellular adhesion molecule-1 (ICAM-1; CD54) expressed on the surface of parenchymal cells (e.g., cardiac myocytes). In the current study, we evaluate the hypothesis that lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18) can also trigger the respiratory burst in neutrophils. To isolate LFA-1/ICAM-1 interactions from Mac-1/ ICAM-1 interactions, full-length chimeric ICAM-1 was developed and expressed in L cells with domains 1 and 2 from canine ICAM-1 and domains 3-5 from human ICAM-1 (C1,2;H3-5). We have shown that canine neutrophils do not bind to human ICAM-1. We demonstrated that chimeric ICAM-1 C1,2;H3-5 supported only LFA-1-dependent adhesion of canine neutrophils and that such adhesion triggered rapid onset of H2O2 production from canine neutrophils. The following seven experimental conditions distinguished LFA-1-dependent H2O2 production from Mac-1-dependent production: It did not require exogenous chemotactic stimulation; H2O2 release was more rapid, but the amount released was <40% of that mediated by Mac-1 adhesion; it was inhibited by anti-CD11a and anti-ICAM-1 antibodies; in contrast to that mediated by Mac-1, it was not inhibited by anti-CD11b antibody, neutrophil inhibitory factor (NIF), or cytochalasin B or H7. Thus, canine neutrophils seem to be able to utilize two members of the beta2 integrin family to interact with ICAM-1 and signal H2O2 production, with LFA-1 at an early stage without prior chemotactic stimulation and Mac-1 at a later stage requiring chemotactic stimulation.
Product Citations
-
Multimodal stimulation screens reveal unique and shared genes limiting T cell fitness.
In Cancer Cell on 8 April 2024 by Lin, C. P., Lévy, P. L., et al.
PubMed
Genes limiting T cell antitumor activity may serve as therapeutic targets. It has not been systematically studied whether there are regulators that uniquely or broadly contribute to T cell fitness. We perform genome-scale CRISPR-Cas9 knockout screens in primary CD8 T cells to uncover genes negatively impacting fitness upon three modes of stimulation: (1) intense, triggering activation-induced cell death (AICD); (2) acute, triggering expansion; (3) chronic, causing dysfunction. Besides established regulators, we uncover genes controlling T cell fitness either specifically or commonly upon differential stimulation. Dap5 ablation, ranking highly in all three screens, increases translation while enhancing tumor killing. Loss of Icam1-mediated homotypic T cell clustering amplifies cell expansion and effector functions after both acute and intense stimulation. Lastly, Ctbp1 inactivation induces functional T cell persistence exclusively upon chronic stimulation. Our results functionally annotate fitness regulators based on their unique or shared contribution to traits limiting T cell antitumor activity.
-
Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH.
In Nature on 1 April 2021 by Dudek, M., Pfister, D., et al.
PubMed
Nonalcoholic steatohepatitis (NASH) is a manifestation of systemic metabolic disease related to obesity, and causes liver disease and cancer1,2. The accumulation of metabolites leads to cell stress and inflammation in the liver3, but mechanistic understandings of liver damage in NASH are incomplete. Here, using a preclinical mouse model that displays key features of human NASH (hereafter, NASH mice), we found an indispensable role for T cells in liver immunopathology. We detected the hepatic accumulation of CD8 T cells with phenotypes that combined tissue residency (CXCR6) with effector (granzyme) and exhaustion (PD1) characteristics. Liver CXCR6+ CD8 T cells were characterized by low activity of the FOXO1 transcription factor, and were abundant in NASH mice and in patients with NASH. Mechanistically, IL-15 induced FOXO1 downregulation and CXCR6 upregulation, which together rendered liver-resident CXCR6+ CD8 T cells susceptible to metabolic stimuli (including acetate and extracellular ATP) and collectively triggered auto-aggression. CXCR6+ CD8 T cells from the livers of NASH mice or of patients with NASH had similar transcriptional signatures, and showed auto-aggressive killing of cells in an MHC-class-I-independent fashion after signalling through P2X7 purinergic receptors. This killing by auto-aggressive CD8 T cells fundamentally differed from that by antigen-specific cells, which mechanistically distinguishes auto-aggressive and protective T cell immunity.
-
Anti-αLβ2 antibodies reveal novel endocytotic cross-modulatory functionality.
In Br J Pharmacol on 1 June 2020 by Mancuso, R. V., Casper, J., et al.
PubMed
Antibodies targeting cell surface receptors are considered to enable highly selective therapeutic interventions for immune disorders and cancer. Their biological profiles are found, generally, to represent the net effects of antibody-target interactions. The former therapeutic anti-integrin αLβ2 antibody efalizumab seems to defeat this paradigm by eliciting, via mechanisms currently unknown, much broader effects than would be predicted based on its target specificity.