InVivoMAb anti-mouse IL-1α
Product Details
The ALF-161 monoclonal antibody reacts with precursor, secreted and membrane-associated forms of mouse IL-1α (interleukin 1 alpha) also known as lymphocyte activating factor (LAF), and mononuclear cell factor (MCF). IL-1α is a 17 kDa pro-inflammatory cytokine produced by a variety of cells, including macrophages, dendritic cells, T and B lymphocytes. IL-1α exerts a wide range of immune and inflammatory responses on a many cell types including lymphocytes, epithelial cells and fibroblasts. IL-1 is made up of IL-1α and IL-1β which are the products of distinct genes, but which are recognized by two distinct IL-1 receptors. The IL-1 receptor type I, a 80 kDa transmembrane protein with demonstrated IL-1 signaling function and the IL-1 receptor type II, a 68 kDa membrane protein with a relatively short cytoplasmic tail. The type II receptor acts as a decoy target for IL-1, inhibiting IL-1 activities by preventing the binding of IL-1 to the type I receptor. The ALF-161 antibody has been shown to neutralize the bioactivity of natural or recombinant IL-1α.Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse IL-1α |
| Reported Applications |
in vivo IL-1α neutralization in vitro IL-1α neutralization |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_2687724 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb polyclonal Armenian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo IL-1α neutralization
Hernández-Santos, N., et al. (2018). "Lung Epithelial Cells Coordinate Innate Lymphocytes and Immunity against Pulmonary Fungal Infection" Cell Host Microbe 23(4): 511-522.e515. PubMed
Lung epithelial cells (LECs) are strategically positioned in the airway mucosa to provide barrier defense. LECs also express pattern recognition receptors and a myriad of immune genes, but their role in immunity is often concealed by the activities of “professional” immune cells, particularly in the context of fungal infection. Here, we demonstrate that NF-κB signaling in LECs is essential for immunity against the pulmonary fungal pathogen Blastomyces dermatitidis. LECs orchestrate innate antifungal immunity by augmenting the numbers of interleukin-17A (IL-17A)- and granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing innate lymphocytes, specifically “natural” Th17 (nTh17) cells. Innate lymphocyte-derived IL-17A and GM-CSF in turn enable phagocyte-driven fungal killing. LECs regulate the numbers of nTh17 cells via the production of chemokines such as CCL20, a process dependent on IL-1α-IL-1 receptor (IL-1R) signaling on LECs. Therefore, LECs orchestrate IL-17A- and GM-CSF-mediated immunity in an IL-1R-dependent manner and represent an essential component of innate immunity to pulmonary fungal pathogens.
in vitro IL-1α neutralization
Altmeier, S., et al. (2016). "IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa" PLoS Pathog 12(9): e1005882. PubMed
Mucosal infections with Candida albicans belong to the most frequent forms of fungal diseases. Host protection is conferred by cellular immunity; however, the induction of antifungal immunity is not well understood. Using a mouse model of oropharyngeal candidiasis (OPC) we show that interleukin-1 receptor (IL-1R) signaling is critical for fungal control at the onset of infection through its impact on neutrophils at two levels. We demonstrate that both the recruitment of circulating neutrophils to the site of infection and the mobilization of newly generated neutrophils from the bone marrow depended on IL-1R. Consistently, IL-1R-deficient mice displayed impaired chemokine production at the site of infection and defective secretion of granulocyte colony-stimulating factor (G-CSF) in the circulation in response to C. albicans. Strikingly, endothelial cells were identified as the primary cellular source of G-CSF during OPC, which responded to IL-1α that was released from keratinocytes in the infected tissue. The IL-1-dependent crosstalk between two different cellular subsets of the nonhematopoietic compartment was confirmed in vitro using a novel murine tongue-derived keratinocyte cell line and an established endothelial cell line. These data establish a new link between IL-1 and granulopoiesis in the context of fungal infection. Together, we identified two complementary mechanisms coordinating the neutrophil response in the oral mucosa, which is critical for preventing fungal growth and dissemination, and thus protects the host from disease.
in vivo IL-1α neutralization
Copenhaver, A. M., et al. (2015). "IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis" Proc Natl Acad Sci U S A 112(24): 7557-7562. PubMed
The innate immune system is critical for host defense against microbial pathogens, yet many pathogens express virulence factors that impair immune function. Here, we used the bacterial pathogen Legionella pneumophila to understand how the immune system successfully overcomes pathogen subversion mechanisms. L. pneumophila replicates within macrophages by using a type IV secretion system to translocate bacterial effectors into the host cell cytosol. As a consequence of effector delivery, host protein synthesis is blocked at several steps, including translation initiation and elongation. Despite this translation block, infected cells robustly produce proinflammatory cytokines, but the basis for this is poorly understood. By using a reporter system that specifically discriminates between infected and uninfected cells within a population, we demonstrate here that infected macrophages produced IL-1alpha and IL-1beta, but were poor producers of IL-6, TNF, and IL-12, which are critical mediators of host protection. Uninfected bystander cells robustly produced IL-6, TNF, and IL-12, and this bystander response required IL-1 receptor (IL-1R) signaling during early pulmonary infection. Our data demonstrate functional heterogeneity in production of critical protective cytokines and suggest that collaboration between infected and uninfected cells enables the immune system to bypass pathogen-mediated translation inhibition to generate an effective immune response.
in vivo IL-1α neutralization
Hernandez, P. P., et al. (2015). "Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection" Nat Immunol 16(7): 698-707. PubMed
The epithelium is the main entry point for many viruses, but the processes that protect barrier surfaces against viral infections are incompletely understood. Here we identified interleukin 22 (IL-22) produced by innate lymphoid cell group 3 (ILC3) as an amplifier of signaling via interferon-lambda (IFN-lambda), a synergism needed to curtail the replication of rotavirus, the leading cause of childhood gastroenteritis. Cooperation between the receptor for IL-22 and the receptor for IFN-lambda, both of which were ‘preferentially’ expressed by intestinal epithelial cells (IECs), was required for optimal activation of the transcription factor STAT1 and expression of interferon-stimulated genes (ISGs). These data suggested that epithelial cells are protected against viral replication by co-option of two evolutionarily related cytokine networks. These data may inform the design of novel immunotherapy for viral infections that are sensitive to interferons.
in vivo IL-1α neutralization
Rogers, H. W., et al. (1992). "Interleukin 1 participates in the development of anti-Listeria responses in normal and SCID mice" Proc Natl Acad Sci U S A 89(3): 1011-1015. PubMed
Using T- and B-cell deficient C.B-17 mice with the scid mutation, we have previously documented the existence of a T-cell-independent but interferon gamma-dependent pathway of macrophage activation that confers upon the host partial resistance to the facultative intracellular bacterium Listeria monocytogenes. This pathway is operative in both normal and SCID mice and consists of at least four components: interferon gamma, tumor necrosis factor, macrophages, and natural killer cells. Here we demonstrate that interleukin 1 also participates in this pathway but at a different site of action. Using monoclonal antibodies that neutralize the biologic activities of interleukin 1 alpha and interleukin 1 beta, we document that interleukin 1 participates neither directly in the induction of interferon gamma from isolated SCID natural killer cells nor in the antigen-specific activation of CD4+ T cells derived from Listeria-immune C.B-17 mice. In contrast, injection of a mixture of anti-interleukin 1 alpha, anti-interleukin 1 beta, and a newly derived monoclonal antibody specific for the murine type I interleukin-1 receptor into either SCID or normal C.B-17 mice blocked the in vivo elaboration of class II major histocompatibility complex-positive macrophages after infection of the animals with Listeria. Moreover, SCID mice treated with the anti-interleukin-1 mixture failed to control the growth of Listeria in vivo and eventually succumbed to the infection. These results document that endogenously produced interleukin 1 plays an obligate role in the Listeria-dependent induction of activated macrophages in vivo and demonstrate that the action of interleukin 1 is distinct from the generation of natural killer cell-derived interferon gamma.
- Immunology and Microbiology,
Fusobacterium nucleatum infection activates the noncanonical inflammasome and exacerbates inflammatory response in DSS-induced colitis.
In European Journal of Immunology on 1 November 2023 by Boonyaleka, K., Okano, T., et al.
PubMed
Caspase activation results in pyroptosis, an inflammatory cell death that contributes to several inflammatory diseases by releasing inflammatory cytokines and cellular contents. Fusobacterium nucleatum is a periodontal pathogen frequently detected in human cancer and inflammatory bowel diseases. Studies have reported that F. nucleatum infection leads to NLRP3 activation and pyroptosis, but the precise activation process and disease association remain poorly understood. This study demonstrated that F. nucleatum infection exacerbates acute colitis in mice and activates pyroptosis through caspase-11-mediated gasdermin D cleavage in macrophages. Furthermore, F. nucleatum infection in colitis mice induces the enhancement of IL-1⍺ secretion from the colon, affecting weight loss and severe disease activities. Neutralization of IL-1⍺ protects F. nucleatum infected mice from severe colitis. Therefore, F. nucleatum infection facilitates inflammation in acute colitis with IL-1⍺ from colon tissue by activating noncanonical inflammasome through gasdermin D cleavage. © 2023 Wiley-VCH GmbH.
IL-1/MyD88-Dependent G-CSF and IL-6 Secretion Mediates Postburn Anemia.
In The Journal of Immunology on 1 April 2023 by Noel, J. G., Ramser, S. W., et al.
PubMed
The anemia of critical illness (ACI) is a nearly universal pathophysiological consequence of burn injury and a primary reason burn patients require massive quantities of transfused blood. Inflammatory processes are expected to drive postburn ACI and prevent meaningful erythropoietic stimulation through iron or erythropoietin supplementation, but to this day no specific inflammatory pathways have been identified as a critical mechanism. In this study, we examined whether secretion of G-CSF and IL-6 mediates distinct features of postburn ACI and interrogated inflammatory mechanisms that could be responsible for their secretion. Our analysis of mouse and human skin samples identified the burn wound as a primary source of G-CSF and IL-6 secretion. We show that G-CSF and IL-6 are secreted independently through an IL-1/MyD88-dependent mechanism, and we ruled out TLR2 and TLR4 as critical receptors. Our results indicate that IL-1/MyD88-dependent G-CSF secretion plays a key role in impairing medullary erythropoiesis and IL-6 secretion plays a key role in limiting the access of erythroid cells to iron. Importantly, we found that IL-1α/β neutralizing Abs broadly attenuated features of postburn ACI that could be attributed to G-CSF or IL-6 secretion and rescued deficits of circulating RBC counts, hemoglobin, and hematocrit caused by burn injury. We conclude that wound-based IL-1/MyD88 signaling mediates postburn ACI through induction of G-CSF and IL-6 secretion. Copyright © 2023 by The American Association of Immunologists, Inc.
- In Vivo,
- Mus musculus (House mouse),
- Immunology and Microbiology,
- Pharmacology
Smoking status impacts treatment efficacy in smoke-induced lung inflammation: A pre-clinical study.
In Frontiers in Pharmacology on 27 September 2022 by Milad, N., Pineault, M., et al.
PubMed
Rationale: Smoking status and smoking history remain poorly accounted for as variables that could affect the efficacy of new drugs being tested in chronic obstructive pulmonary disease (COPD) patients. As a proof of concept, we used a pre-clinical model of cigarette smoke (CS) exposure to compare the impact of treatment during active CS exposure or during the cessation period on the anti-inflammatory effects IL-1α signaling blockade. Methods: Mice were exposed to CS for 2 weeks, followed by a 1-week cessation, then acutely re-exposed for 2 days. Mice were treated with an anti-IL-1α antibody either during CS exposure or during cessation and inflammatory outcomes were assessed. Results: We found that mice re-exposed to CS displayed reduced neutrophil counts and cytokine levels in the bronchoalveolar lavage (BAL) compared to mice exposed only acutely. Moreover, we found that treatment with an anti-IL-1α antibody during the initial CS exposure delayed inflammatory processes and interfered with pulmonary adaptation, leading to rebound pulmonary neutrophilia, increased BAL cytokine secretion (CCL2) and upregulated Mmp12 expression. Conversely, administration of anti-IL-1α during cessation had the opposite effect, improving BAL neutrophilia, decreasing CCL2 levels and reducing Mmp12 expression. Discussion: These results suggest that pulmonary adaptation to CS exposure dampens inflammation and blocking IL-1α signaling during CS exposure delays the inflammatory response. More importantly, the same treatment administered during cessation hastens the return to pulmonary inflammatory homeostasis, strongly suggesting that smoking status and treatment timing should be considered when testing new biologics in COPD. Copyright © 2022 Milad, Pineault, Tremblay, Routhier, Lechasseur, Beaulieu, Aubin and Morissette.
- In Vivo,
- Mus musculus (House mouse),
- Neuroscience
Interleukin-1 Is Overexpressed in Injured Muscles Following Spinal Cord Injury and Promotes Neurogenic Heterotopic Ossification.
In Journal of Bone and Mineral Research on 1 March 2022 by Tseng, H. W., Kulina, I., et al.
PubMed
Neurogenic heterotopic ossifications (NHOs) form in periarticular muscles after severe spinal cord (SCI) and traumatic brain injuries. The pathogenesis of NHO is poorly understood with no effective preventive treatment. The only curative treatment remains surgical resection of pathological NHOs. In a mouse model of SCI-induced NHO that involves a transection of the spinal cord combined with a muscle injury, a differential gene expression analysis revealed that genes involved in inflammation such as interleukin-1β (IL-1β) were overexpressed in muscles developing NHO. Using mice knocked-out for the gene encoding IL-1 receptor (IL1R1) and neutralizing antibodies for IL-1α and IL-1β, we show that IL-1 signaling contributes to NHO development after SCI in mice. Interestingly, other proteins involved in inflammation that were also overexpressed in muscles developing NHO, such as colony-stimulating factor-1, tumor necrosis factor, or C-C chemokine ligand-2, did not promote NHO development. Finally, using NHO biopsies from SCI and TBI patients, we show that IL-1β is expressed by CD68+ macrophages. IL-1α and IL-1β produced by activated human monocytes promote calcium mineralization and RUNX2 expression in fibro-adipogenic progenitors isolated from muscles surrounding NHOs. Altogether, these data suggest that interleukin-1 promotes NHO development in both humans and mice. © 2021 American Society for Bone and Mineral Research (ASBMR). © 2021 American Society for Bone and Mineral Research (ASBMR).
Pathogenic, but not non-pathogenic, i>Rickettsia/i> evade inflammasome-dependent IL-1 responses to establish an intracytosolic replication niche
Preprint on BioRxiv : the Preprint Server for Biology on 8 September 2021 by Voss, O. H., Cobb, J., et al.
PubMed
h4>ABSTRACT/h4> Rickettsia species (spp.) are strict obligate intracellular bacteria, with some being pathogenic in their mammalian host, including humans. One critical feature of these stealthy group of pathogens is their ability to manipulate hostile cytosolic environments to their benefits. Although our understanding of Rickettsia cell biology and pathogenesis are evolving, the mechanisms by which pathogenic Rickettsia spp. evade host innate immune detection remains elusive. Here, we showed that disease severity in wild-type ( WT ) C57BL/6J mice infected with R. typhi (etiologic agent of murine typhus ) and R. rickettsii (etiologic agent of Rocky Mountain Spotted Fever), but not with non-pathogenic R. montanensis , correlated with levels of bacterial burden as detected in the spleens, as well as the serum concentrations of pro-inflammatory cytokine IL-1α and to a lesser extent IL- 1β. Antibody-mediated neutralization of IL-1α confirmed a key role in controlling mortality rates and bacterial burdens of rickettsiae-infected WT mice. As macrophages are a primary source of both IL-1α and IL-1β cytokines, we determined the mechanism of the anti-rickettsial activities using bone-marrow-derived macrophages. We found that pathogenic R. typhi and R. rickettsii , but not non-pathogenic R. montanensis , eluded pro- IL-1α induction and benefited pre-dominantly from the reduced IL-1α secretion, via a Caspase-11-Gsdmd-dependent pathway, to facilitate intracytosolic replication. Adoptative transfer experiments identified that IL-1α secretion by macrophages was critical for controlling rickettsiosis in WT mice. In sum, we identified a previously unappreciated pathway by which pathogenic, unlike non-pathogenic, rickettsiae preferentially target the Caspase-11-Gsdmd-IL-1α signaling axis in macrophages thus supporting their replication within the host. h4>IMPORTANCE/h4> Currently, no vaccines are available to prevent rickettsioses, while vector-borne rickettsial infections in humans are on the rise globally. In fact, the insufficient understanding of how pathogenic Rickettsia species circumvent host immune defense mechanisms has significantly hindered the development of more effective therapeutics. Here, we identified a previously unappreciated role for the Caspase-11-Gsdmd-IL-1α signaling axis, to limiting the replication of pathogenic R. rickettsia and R. typhi species in murine macrophages and wild-type ( WT ) C57BL/6J mice. Adoptative transfer studies further identified IL-1α-secreting macrophages as critical mediators in controlling rickettsial infection in WT mice. Collectively, these findings provide insight into the potential mechanism of how pathogenic, but not non-pathogenic Rickettsia spp., benefit from a reduction in the Caspase-11-Gsdmd-mediated release of IL-1α to support host colonization.
- In Vivo,
- Mus musculus (House mouse),
- Immunology and Microbiology
Neutrophils and IL-1α Regulate Surfactant Homeostasis during Cigarette Smoking.
In The Journal of Immunology on 15 April 2021 by Milad, N., Pineault, M., et al.
PubMed
Cigarette smoke exposure induces inflammation marked by rapid and sustained neutrophil infiltration, IL-1α, release and altered surfactant homeostasis. However, the extent to which neutrophils and IL-1α contribute to the maintenance of pulmonary surfactant homeostasis is not well understood. We sought to investigate whether neutrophils play a role in surfactant clearance as well as the effect of neutrophil depletion and IL-1α blockade on the response to cigarette smoke exposure. In vitro and in vivo administration of fluorescently labeled surfactant phosphatidylcholine was used to assess internalization of surfactant by lung neutrophils and macrophages during or following cigarette smoke exposure in mice. We also depleted neutrophils using anti-Ly-6G or anti-Gr-1 Abs, or we neutralized IL-1α using a blocking Ab to determine their respective roles in regulating surfactant homeostasis during cigarette smoke exposure. We observed that neutrophils actively internalize labeled surfactant both in vitro and in vivo and that IL-1α is required for smoke-induced elevation of surfactant protein (SP)-A and SP-D levels. Neutrophil depletion during cigarette smoke exposure led to a further increase in SP-A levels in the bronchoalveolar lavage and increased IL-1α, CCL2, GM-CSF, and G-CSF release. Finally, macrophage expression of Mmp12, a protease linked to emphysema, was increased in neutrophil-depleted groups and decreased following IL-1α blockade. Taken together, our results indicate that neutrophils and IL-1α signaling are actively involved in surfactant homeostasis and that the absence of neutrophils in the lungs during cigarette smoke exposure leads to an IL-1α-dependent exacerbation of the inflammatory response.Copyright © 2021 by The American Association of Immunologists, Inc.
- In Vivo,
- Mus musculus (House mouse)
Pathogenic, but Not Nonpathogenic, Rickettsia spp. Evade Inflammasome-Dependent IL-1 Responses To Establish an Intracytosolic Replication Niche.
In mBio on 22 February 2021 by Voss, O. H., Cobb, J., et al.
PubMed
Rickettsia species (spp.) are strict obligate intracellular bacteria, some of which are pathogenic in their mammalian host, including humans. One critical feature of these stealthy group of pathogens is their ability to manipulate hostile cytosolic environments to their benefits. Although our understanding of Rickettsia cell biology and pathogenesis is evolving, the mechanisms by which pathogenic Rickettsia spp. evade host innate immune detection remain elusive. Here, we show that disease severity in wild-type (WT) C57BL/6J mice infected with Rickettsia typhi (the etiologic agent of murine typhus) and Rickettsia rickettsii (the etiologic agent of Rocky Mountain spotted fever), but not with the nonpathogenic species Rickettsia montanensis, correlated with levels of bacterial burden as detected in the spleens of mice, as well as the serum concentrations of proinflammatory cytokine interleukin-1α (IL-1α) and, to a lesser extent, IL-1β. Antibody-mediated neutralization of IL-1α confirmed a key role in controlling mortality rates and bacterial burdens of rickettsia-infected WT mice. As macrophages are a primary source of both IL-1α and IL-1β cytokines, we determined the mechanism of the antirickettsial activities using bone marrow-derived macrophages. We found that pathogenic R. typhi and R. rickettsii, but not nonpathogenic R. montanensis, eluded pro-IL-1α induction and benefited predominantly from the reduced IL-1α secretion, via a caspase-11-gasdermin D (Gsdmd)-dependent pathway, to facilitate intracytosolic replication. Adoptive transfer experiments identified that IL-1α secretion by macrophages was critical for controlling rickettsiosis in WT mice. In sum, we identified a previously unappreciated pathway by which pathogenic, unlike nonpathogenic, rickettsiae preferentially target the caspase-11-Gsdmd-IL-1α signaling axis in macrophages, thus supporting their replication within the host. IMPORTANCE Currently, no vaccines are available to prevent rickettsioses, while vector-borne rickettsial infections in humans are on the rise globally. In fact, the insufficient understanding of how pathogenic Rickettsia species circumvent host immune defense mechanisms has significantly hindered the development of more effective therapeutics. Here, we identified a previously unappreciated role for the caspase-11-Gsdmd-IL-1α signaling axis in limiting the replication of pathogenic R. rickettsia and R. typhi species in murine macrophages and wild-type (WT) C57BL/6J mice. Adoptive transfer studies further identified IL-1α-secreting macrophages as critical mediators in controlling rickettsial infection in WT mice. Collectively, these findings provide insight into the potential mechanism of how pathogenic, but not nonpathogenic, Rickettsia spp. benefit from a reduction in the caspase-11-Gsdmd-mediated release of IL-1α to support host colonization.
- In Vivo,
- Mus musculus (House mouse),
- Cancer Research,
- Immunology and Microbiology
Systemic dysfunction and plasticity of the immune macroenvironment in cancer models.
In Nature Medicine on 1 July 2020 by Allen, B. M., Hiam, K. J., et al.
PubMed
Understanding of the factors governing immune responses in cancer remains incomplete, limiting patient benefit. In this study, we used mass cytometry to define the systemic immune landscape in response to tumor development across five tissues in eight mouse tumor models. Systemic immunity was dramatically altered across models and time, with consistent findings in the peripheral blood of patients with breast cancer. Changes in peripheral tissues differed from those in the tumor microenvironment. Mice with tumor-experienced immune systems mounted dampened responses to orthogonal challenges, including reduced T cell activation during viral or bacterial infection. Antigen-presenting cells (APCs) mounted weaker responses in this context, whereas promoting APC activation rescued T cell activity. Systemic immune changes were reversed with surgical tumor resection, and many were prevented by interleukin-1 or granulocyte colony-stimulating factor blockade, revealing remarkable plasticity in the systemic immune state. These results demonstrate that tumor development dynamically reshapes the composition and function of the immune macroenvironment.
- Cell Biology,
- Immunology and Microbiology
Autophagy Protects Against Developing Increased Lung Permeability and Hypoxemia by Down Regulating Inflammasome Activity and IL-1β in LPS Plus Mechanical Ventilation-Induced Acute Lung Injury.
In Frontiers in Immunology on 3 March 2020 by Nosaka, N., Martinon, D., et al.
PubMed
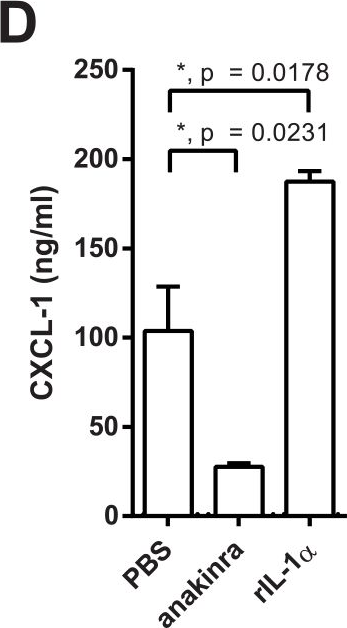
Targeting inflammasome activation to modulate interleukin (IL)-1β is a promising treatment strategy against acute respiratory distress syndrome and ventilator-induced lung injury (VILI). Autophagy is a key regulator of inflammasome activation in macrophages. Here, we investigated the role of autophagy in the development of acute lung injury (ALI) induced by lipopolysaccharide (LPS) and mechanical ventilation (MV). Two hours before starting MV, 0.2 mg/kg LPS was administered to mice intratracheally. Mice were then placed on high-volume MV (30 ml/kg with 3 cmH2O positive end-expiratory pressure for 2.5 h without additional oxygen application). Mice with myeloid-specific deletion of the autophagic protein ATG16L1 (Atg16l1fl/flLysMCre) suffered severe hypoxemia (adjusted p 0.05) and increased lung permeability (p 0.05, albumin level in bronchoalveolar lavage fluid) with significantly higher IL-1β release into alveolar space (p 0.05). Induction of autophagy by fasting-induced starvation led to improved arterial oxygenation (adjusted p 0.0001) and lung permeability (p 0.05), as well as significantly suppressed IL-1β production (p 0.01). Intratracheal treatment with anti-mouse IL-1β monoclonal antibody (mAb; 2.5 mg/kg) significantly improved arterial oxygenation (adjusted p 0.01) as well as lung permeability (p 0.05). On the other hand, deletion of IL-1α gene or use of anti-mouse IL-1α mAb (2.5 mg/kg) provided no significant protection, suggesting that the LPS and MV-induced ALI is primarily dependent on IL-1β, but independent of IL-1α. These observations suggest that autophagy has a protective role in controlling inflammasome activation and production of IL-1β, which plays a critical role in developing hypoxemia and increased lung permeability in LPS plus MV-induced acute lung injury. Copyright © 2020 Nosaka, Martinon, Moreira, Crother, Arditi and Shimada.
- Neutralization,
- Mus musculus (House mouse),
- Cardiovascular biology,
- Immunology and Microbiology
Lung Epithelial Cells Coordinate Innate Lymphocytes and Immunity against Pulmonary Fungal Infection.
In Cell Host & Microbe on 11 April 2018 by Hernández-Santos, N., Wiesner, D. L., et al.
PubMed
Lung epithelial cells (LECs) are strategically positioned in the airway mucosa to provide barrier defense. LECs also express pattern recognition receptors and a myriad of immune genes, but their role in immunity is often concealed by the activities of "professional" immune cells, particularly in the context of fungal infection. Here, we demonstrate that NF-κB signaling in LECs is essential for immunity against the pulmonary fungal pathogen Blastomyces dermatitidis. LECs orchestrate innate antifungal immunity by augmenting the numbers of interleukin-17A (IL-17A)- and granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing innate lymphocytes, specifically "natural" Th17 (nTh17) cells. Innate lymphocyte-derived IL-17A and GM-CSF in turn enable phagocyte-driven fungal killing. LECs regulate the numbers of nTh17 cells via the production of chemokines such as CCL20, a process dependent on IL-1α-IL-1 receptor (IL-1R) signaling on LECs. Therefore, LECs orchestrate IL-17A- and GM-CSF-mediated immunity in an IL-1R-dependent manner and represent an essential component of innate immunity to pulmonary fungal pathogens. Copyright © 2018 Elsevier Inc. All rights reserved.
- In Vivo,
- Immu-depl,
- Mus musculus (House mouse),
- Immunology and Microbiology
Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation.
In Cell Host & Microbe on 8 November 2017 by Nakagawa, S., Matsumoto, M., et al.
PubMed
Staphylococcus aureus commonly colonizes the epidermis, but the mechanisms by which the host senses virulent, but not commensal, S. aureus to trigger inflammation remain unclear. Using a murine epicutaneous infection model, we found that S. aureus-expressed phenol-soluble modulin (PSM)α, a group of secreted virulence peptides, is required to trigger cutaneous inflammation. PSMα induces the release of keratinocyte IL-1α and IL-36α, and signaling via IL-1R and IL-36R was required for induction of the pro-inflammatory cytokine IL-17. The levels of released IL-1α and IL-36α, as well as IL-17 production by γδ T cells and ILC3 and neutrophil infiltration to the site of infection, were greatly reduced in mice with total or keratinocyte-specific deletion of the IL-1R and IL-36R signaling adaptor Myd88. Further, Il17a-/-f-/- mice showed blunted S. aureus-induced inflammation. Thus, keratinocyte Myd88 signaling in response to S. aureus PSMα drives an IL-17-mediated skin inflammatory response to epicutaneous S. aureus infection. Copyright © 2017 Elsevier Inc. All rights reserved.
- IHC-Fr-IF,
- Mus musculus (House mouse),
- Immunology and Microbiology
The intraspecies diversity of C. albicans triggers qualitatively and temporally distinct host responses that determine the balance between commensalism and pathogenicity.
In Mucosal Immunology on 1 September 2017 by Schönherr, F. A., Sparber, F., et al.
PubMed
The host immune status is critical for preventing opportunistic infections with Candida albicans. Whether the natural fungal diversity that exists between C. albicans isolates also influences disease development remains unclear. Here, we used an experimental model of oral infection to probe the host response to diverse C. albicans isolates in vivo and found dramatic differences in their ability to persist in the oral mucosa, which inversely correlated with the degree and kinetics of immune activation in the host. Strikingly, the requirement of interleukin (IL)-17 signaling for fungal control was conserved between isolates, including isolates with delayed induction of IL-17. This underscores the relevance of IL-17 immunity in mucosal defense against C. albicans. In contrast, the accumulation of neutrophils and induction of inflammation in the infected tissue was strictly strain dependent. The dichotomy of the inflammatory neutrophil response was linked to the capacity of fungal strains to cause cellular damage and release of alarmins from the epithelium. The epithelium thus translates differences in the fungus into qualitatively distinct host responses. Altogether, this study provides a comprehensive understanding of the antifungal response in the oral mucosa and demonstrates the relevance of evaluating intraspecies differences for the outcome of fungal-host interactions in vivo.
- ELISA,
- Mus musculus (House mouse),
- Immunology and Microbiology
IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa.
In PLoS Pathogens on 1 September 2016 by Altmeier, S., Toska, A., et al.
PubMed
Mucosal infections with Candida albicans belong to the most frequent forms of fungal diseases. Host protection is conferred by cellular immunity; however, the induction of antifungal immunity is not well understood. Using a mouse model of oropharyngeal candidiasis (OPC) we show that interleukin-1 receptor (IL-1R) signaling is critical for fungal control at the onset of infection through its impact on neutrophils at two levels. We demonstrate that both the recruitment of circulating neutrophils to the site of infection and the mobilization of newly generated neutrophils from the bone marrow depended on IL-1R. Consistently, IL-1R-deficient mice displayed impaired chemokine production at the site of infection and defective secretion of granulocyte colony-stimulating factor (G-CSF) in the circulation in response to C. albicans. Strikingly, endothelial cells were identified as the primary cellular source of G-CSF during OPC, which responded to IL-1α that was released from keratinocytes in the infected tissue. The IL-1-dependent crosstalk between two different cellular subsets of the nonhematopoietic compartment was confirmed in vitro using a novel murine tongue-derived keratinocyte cell line and an established endothelial cell line. These data establish a new link between IL-1 and granulopoiesis in the context of fungal infection. Together, we identified two complementary mechanisms coordinating the neutrophil response in the oral mucosa, which is critical for preventing fungal growth and dissemination, and thus protects the host from disease.
IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis.
In Proceedings of the National Academy of Sciences of the United States of America on 16 June 2015 by Copenhaver, A. M., Casson, C. N., et al.
PubMed
The innate immune system is critical for host defense against microbial pathogens, yet many pathogens express virulence factors that impair immune function. Here, we used the bacterial pathogen Legionella pneumophila to understand how the immune system successfully overcomes pathogen subversion mechanisms. L. pneumophila replicates within macrophages by using a type IV secretion system to translocate bacterial effectors into the host cell cytosol. As a consequence of effector delivery, host protein synthesis is blocked at several steps, including translation initiation and elongation. Despite this translation block, infected cells robustly produce proinflammatory cytokines, but the basis for this is poorly understood. By using a reporter system that specifically discriminates between infected and uninfected cells within a population, we demonstrate here that infected macrophages produced IL-1α and IL-1β, but were poor producers of IL-6, TNF, and IL-12, which are critical mediators of host protection. Uninfected bystander cells robustly produced IL-6, TNF, and IL-12, and this bystander response required IL-1 receptor (IL-1R) signaling during early pulmonary infection. Our data demonstrate functional heterogeneity in production of critical protective cytokines and suggest that collaboration between infected and uninfected cells enables the immune system to bypass pathogen-mediated translation inhibition to generate an effective immune response.