InVivoMAb anti-mouse VEGF-A
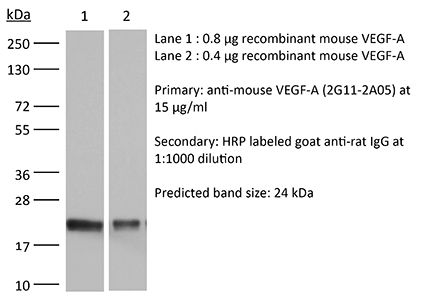
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | N-terminal 24 AA sequence of murine VEGF |
| Reported Applications |
in vivo VEGF-A neutralization Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Kaneko H, Kuroshima S, Kozutsumi R, Al-Omari FA, Hayano H, Nakajima K, Sawase T (2023). "Zoledronate/Anti-VEGF Neutralizing Antibody Combination Administration Increases Osteal Macrophages in a Murine Model of MRONJ Stage 0-like Lesions" J Clin Med 1
PubMed
The pathophysiology, pathogenesis, histopathology, and immunopathology of medication-related osteonecrosis of the jaw (MRONJ) Stage 0 remain unclear, although 50% of MRONJ Stage 0 cases could progress to higher stages. The aim of this study was to investigate the effects of zoledronate (Zol) and anti-vascular endothelial cell growth factor A (VEGFA) neutralizing antibody (Vab) administration on polarization shifting of macrophage subsets in tooth extraction sockets by creating a murine model of MRONJ Stage 0-like lesions. Eight-week-old, female C57BL/6J mice were randomly divided into 4 groups: Zol, Vab, Zol/Vab combination, and vehicle control (VC). Subcutaneous Zol and intraperitoneal Vab administration were performed for 5 weeks with extraction of both maxillary first molars 3 weeks after drug administration. Euthanasia was conducted 2 weeks after tooth extraction. Maxillae, tibiae, femora, tongues, and sera were collected. Structural, histological, immunohistochemical, and biochemical analyses were comprehensively performed. Tooth extraction sites appeared to be completely healed in all groups. However, osseous healing and soft tissue healing of tooth extraction sites were quite different. The Zol/Vab combination significantly induced abnormal epithelial healing, and delayed connective tissue healing due to decreased rete ridge length and thickness of the stratum granulosum and due to decreased collagen production, respectively. Moreover, Zol/Vab significantly increased necrotic bone area with increased numbers of empty lacunae compared with Vab and VC. Most interestingly, Zol/Vab significantly increased the number of CD169+ osteal macrophages (osteomacs) in the bone marrow and decreased F4/80+ macrophages, with a slightly increased ratio of F4/80+CD38+ M1 macrophages compared to VC. These findings are the first to provide new evidence of the involvement of osteal macrophages in the immunopathology of MRONJ Stage 0-like lesions.
-
Mashima T, Wakatsuki T, Kawata N, Jang MK, Nagamori A, Yoshida H, Nakamura K, Migita T, Seimiya H, Yamaguchi K (2021). "Neutralization of the induced VEGF-A potentiates the therapeutic effect of an anti-VEGFR2 antibody on gastric cancer in vivo" Sci
PubMed
The vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) axis is an essential regulator of angiogenesis and important therapeutic target in cancer. Ramucirumab is an anti-VEGFR2 monoclonal antibody used for the treatment of several cancers. Increased circulating VEGF-A levels after ramucirumab administration are associated with a worse prognosis, suggesting that excess VEGF-A induced by ramucirumab negatively affects treatment efficacy and that neutralizing VEGF-A may improve treatment outcomes. Here, we evaluated the effect of combination treatment with an anti-VEGFR2 antibody and anti-VEGF-A antibody on gastric tumor progression and normal tissues using a preclinical BALB/c-nu/nu mouse xenograft model. After anti-VEGFR2 antibody treatment in mice, a significant increase in plasma VEGF-A levels was observed, mirroring the clinical response. The elevated VEGF-A was host-derived. Anti-VEGF-A antibody co-administration enhanced the anti-tumor effect of the anti-VEGFR2-antibody without exacerbating the toxicity. Mechanistically, the combination treatment induced intra-tumor molecular changes closely related to angiogenesis inhibition and abolished the gene expression changes specifically induced by anti-VEGFR2 antibody treatment alone. We particularly identified the dual treatment-selective downregulation of ZEB1 expression, which was critical for gastric cancer cell proliferation. These data indicate that the dual blockade of VEGF-A and VEGFR2 is a rational strategy to ensure the anti-tumor effect of angiogenesis-targeting therapy.
-
Huang J, Kelly CP, Bakirtzi K, Villafuerte Gálvez JA, Lyras D, Mileto SJ, Larcombe S, Xu H, Yang X, Shields KS, Zhu W, Zhang Y, Goldsmith JD, Patel IJ, Hansen J, Huang M, Yla-Herttuala S, Moss AC, Paredes-Sabja D, Pothoulakis C, Shah YM, Wang J, Chen
PubMed
Clostridium difficile infection (CDI) is mediated by two major exotoxins, toxin A (TcdA) and toxin B (TcdB), that damage the colonic epithelial barrier and induce inflammatory responses. The function of the colonic vascular barrier during CDI has been relatively understudied. Here we report increased colonic vascular permeability in CDI mice and elevated vascular endothelial growth factor A (VEGF-A), which was induced in vivo by infection with TcdA- and/or TcdB-producing C. difficile strains but not with a TcdA-TcdB- isogenic mutant. TcdA or TcdB also induced the expression of VEGF-A in human colonic mucosal biopsies. Hypoxia-inducible factor signalling appeared to mediate toxin-induced VEGF production in colonocytes, which can further stimulate human intestinal microvascular endothelial cells. Both neutralization of VEGF-A and inhibition of its signalling pathway attenuated CDI in vivo. Compared to healthy controls, CDI patients had significantly higher serum VEGF-A that subsequently decreased after treatment. Our findings indicate critical roles for toxin-induced VEGF-A and colonic vascular permeability in CDI pathogenesis and may also point to the pathophysiological significance of the gut vascular barrier in response to virulence factors of enteric pathogens. As an alternative to pathogen-targeted therapy, this study may enable new host-directed therapeutic approaches for severe, refractory CDI.
-
Kumar V, Scandella E, Danuser R, Onder L, Nitschké M, Fukui Y, Halin C, Ludewig B, Stein JV (2010). "Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway" Blood 115(23):4725-33.
PubMed
Adaptive immune responses are characterized by substantial restructuring of secondary lymphoid organs. The molecular and cellular factors responsible for virus-induced lymphoid remodeling are not well known to date. Here we applied optical projection tomography, a mesoscopic imaging technique, for a global analysis of the entire 3-dimensional structure of mouse peripheral lymph nodes (PLNs), focusing on B-cell areas and high endothelial venule (HEV) networks. Structural homeostasis of PLNs was characterized by a strict correlation between total PLN volume, B-cell volume, B-cell follicle number, and HEV length. After infection with lymphocytic choriomeningitis virus, we observed a substantial, lymphotoxin (LT) beta-receptor-dependent reorganization of the PLN microarchitecture, in which an initial B-cell influx was followed by 3-fold increases in PLN volume and HEV network length on day 8 after infection. Adoptive transfer experiments revealed that virus-induced PLN and HEV network remodeling required LTalpha(1)beta(2)-expressing B cells, whereas the inhibition of vascular endothelial growth factor-A signaling pathways had no significant effect on PLN expansion. In summary, lymphocytic choriomeningitis virus-induced PLN growth depends on a vascular endothelial growth factor-A-independent, LT- and B cell-dependent morphogenic pathway, as revealed by an in-depth mesoscopic analysis of the global PLN structure.
Product Citations
-
Aspergillus fumigatus promotes tumor angiogenesis via SLC7A11 on myeloid-derived suppressor cells.
In EMBO Rep on 1 December 2025 by Qu, W., Wang, Z., et al.
PubMed
The microbiome is increasingly recognized as playing a critical role in lung cancer prevention, diagnosis, and treatment. While bacteria are essential for tumor angiogenesis, the impact of fungi on this process remains largely unexplored. In this study, we investigate effects of Aspergillus fumigatus (A. fumigatus) on lung cancer. We show that inhalation of A. fumigatus increases tumor burden and angiogenesis in mouse models. Interestingly, A. fumigatus does not directly affect the proangiogenic abilities of tumor cells or endothelial cells. Instead, A. fumigatus promotes the accumulation of myeloid-derived suppressor cells (MDSCs), particularly G-MDSCs, in tumor tissues. A. fumigatus increases VEGF-A secretion from tumor-associated MDSCs, promoting tumor angiogenesis. Furthermore, we identify solute carrier family 7 member 11 (SLC7A11) as a key player in regulating this proangiogenic function through an interaction with High Mobility Group Box 1 (HMGB1) in MDSCs. Our results shed light on the mechanisms by which A. fumigatus influences MDSCs to promote angiogenesis and demonstrate that commensal fungi influence host immunity and support tumor progression.
-
Serotonin receptor 5-HT2A as a potential target for HCC immunotherapy.
In J Immunother Cancer on 23 June 2025 by Tay, R. E., Ho, C. M., et al.
PubMed
While recent clinical trials of combination immunotherapies for hepatocellular carcinoma (HCC) have shown promising clinical efficacy and survival improvements breakthroughs, there is still much room for further improvement. A key limiting factor for HCC immunotherapy is the intrinsic immunosuppression within the liver microenvironment, resulting in suboptimal priming of tumor-specific CD8 cytotoxic T cells and thus immune evasion by the tumor. Hence, identifying new key molecular pathways suppressing T-cell responses within the liver is critical for the rational design of more effective combination immunotherapies for HCC.