InVivoMAb anti-mouse MHC Class I (H-2)
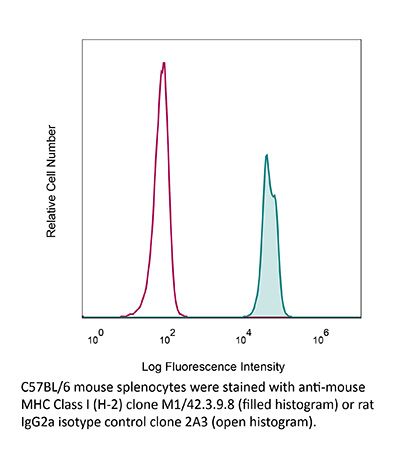
Product Description
Specifications
| Isotype | Rat IgG2a, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2a isotype control, anti-trinitrophenol |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | C57BL/10 mouse spleen cells enriched for T cells |
| Reported Applications |
ex vivo blocking of MHC I-dependent interactions MHC-I immunopeptidomics Immunoprecipitation Flow cytometry Immunofluorescence |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1125537 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Herz, J., et al (2015). "Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells" J Exp Med 212(8): 1153-1169.
PubMed
Several viruses can infect the mammalian nervous system and induce neurological dysfunction. Adoptive immunotherapy is an approach that involves administration of antiviral T cells and has shown promise in clinical studies for the treatment of peripheral virus infections in humans such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and adenovirus, among others. In contrast, clearance of neurotropic infections is particularly challenging because the central nervous system (CNS) is relatively intolerant of immunopathological reactions. Therefore, it is essential to develop and mechanistically understand therapies that noncytopathically eradicate pathogens from the CNS. Here, we used mice persistently infected from birth with lymphocytic choriomeningitis virus (LCMV) to demonstrate that therapeutic antiviral T cells can completely purge the persistently infected brain without causing blood-brain barrier breakdown or tissue damage. Mechanistically, this is accomplished through a tailored release of chemoattractants that recruit antiviral T cells, but few pathogenic innate immune cells such as neutrophils and inflammatory monocytes. Upon arrival, T cells enlisted the support of nearly all brain-resident myeloid cells (microglia) by inducing proliferation and converting them into CD11c(+) antigen-presenting cells (APCs). Two-photon imaging experiments revealed that antiviral CD8(+) and CD4(+) T cells interacted directly with CD11c(+) microglia and induced STAT1 signaling but did not initiate programmed cell death. We propose that noncytopathic CNS viral clearance can be achieved by therapeutic antiviral T cells reliant on restricted chemoattractant production and interactions with apoptosis-resistant microglia.
-
Rockett, B. D., et al (2011). "Membrane raft organization is more sensitive to disruption by (n-3) PUFA than nonraft organization in EL4 and B cells" J Nutr 141(6): 1041-1048.
PubMed
Model membrane and cellular detergent extraction studies show (n-3) PUFA predominately incorporate into nonrafts; thus, we hypothesized (n-3) PUFA could disrupt nonraft organization. The first objective of this study was to determine whether (n-3) PUFA disrupted nonrafts of EL4 cells, an extension of our previous work in which we discovered an (n-3) PUFA diminished raft clustering. EPA or DHA treatment of EL4 cells increased plasma membrane accumulation of the nonraft probe 1,1′-dilinoleyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate by ~50-70% relative to a BSA control. Forster resonance energy transfer imaging showed EPA and DHA also disrupted EL4 nanometer scale nonraft organization by increasing the distance between nonraft molecules by ~25% compared with BSA. However, changes in nonrafts were due to an increase in cell size; under conditions where EPA or DHA did not increase cell size, nonraft organization was unaffected. We next translated findings on EL4 cells by testing if (n-3) PUFA administered to mice disrupted nonrafts and rafts. Imaging of B cells isolated from mice fed low- or high-fat (HF) (n-3) PUFA diets showed no change in nonraft organization compared with a control diet (CD). However, confocal microscopy revealed the HF (n-3) PUFA diet disrupted lipid raft clustering and size by ~40% relative to CD. Taken together, our data from 2 different model systems suggest (n-3) PUFA have limited effects on nonrafts. The ex vivo data, which confirm previous studies with EL4 cells, provide evidence that (n-3) PUFA consumed through the diet disrupt B cell lipid raft clustering.
-
Shaikh, S. R., et al (2009). "Docosahexaenoic acid modifies the clustering and size of lipid rafts and the lateral organization and surface expression of MHC class I of EL4 cells" J Nutr 139(9): 1632-1639.
PubMed
An emerging molecular mechanism by which docosahexaenoic acid (DHA) exerts its effects is modification of lipid raft organization. The biophysical model, based on studies with liposomes, shows that DHA avoids lipid rafts because of steric incompatibility between DHA and cholesterol. The model predicts that DHA does not directly modify rafts; rather, it incorporates into nonrafts to modify the lateral organization and/or conformation of membrane proteins, such as the major histocompatibility complex (MHC) class I. Here, we tested predictions of the model at a cellular level by incorporating oleic acid, eicosapentaenoic acid (EPA), and DHA, compared with a bovine serum albumin (BSA) control, into the membranes of EL4 cells. Quantitative microscopy showed that DHA, but not EPA, treatment, relative to the BSA control diminished lipid raft clustering and increased their size. Approximately 30% of DHA was incorporated directly into rafts without changing the distribution of cholesterol between rafts and nonrafts. Quantification of fluorescence colocalization images showed that DHA selectively altered MHC class I lateral organization by increasing the fraction of the nonraft protein into rafts compared with BSA. Both DHA and EPA treatments increased antibody binding to MHC class I compared with BSA. Antibody titration showed that DHA and EPA did not change MHC I conformation but increased total surface levels relative to BSA. Taken together, our findings are not in agreement with the biophysical model. Therefore, we propose a model that reconciles contradictory viewpoints from biophysical and cellular studies to explain how DHA modifies lipid rafts on several length scales. Our study supports the notion that rafts are an important target of DHA’s mode of action.
-
Herz, J., et al (2015). "Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells" J Exp Med 212(8): 1153-1169.
PubMed
Several viruses can infect the mammalian nervous system and induce neurological dysfunction. Adoptive immunotherapy is an approach that involves administration of antiviral T cells and has shown promise in clinical studies for the treatment of peripheral virus infections in humans such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and adenovirus, among others. In contrast, clearance of neurotropic infections is particularly challenging because the central nervous system (CNS) is relatively intolerant of immunopathological reactions. Therefore, it is essential to develop and mechanistically understand therapies that noncytopathically eradicate pathogens from the CNS. Here, we used mice persistently infected from birth with lymphocytic choriomeningitis virus (LCMV) to demonstrate that therapeutic antiviral T cells can completely purge the persistently infected brain without causing blood-brain barrier breakdown or tissue damage. Mechanistically, this is accomplished through a tailored release of chemoattractants that recruit antiviral T cells, but few pathogenic innate immune cells such as neutrophils and inflammatory monocytes. Upon arrival, T cells enlisted the support of nearly all brain-resident myeloid cells (microglia) by inducing proliferation and converting them into CD11c(+) antigen-presenting cells (APCs). Two-photon imaging experiments revealed that antiviral CD8(+) and CD4(+) T cells interacted directly with CD11c(+) microglia and induced STAT1 signaling but did not initiate programmed cell death. We propose that noncytopathic CNS viral clearance can be achieved by therapeutic antiviral T cells reliant on restricted chemoattractant production and interactions with apoptosis-resistant microglia.
Product Citations
-
Oncolytic virus M1 reinvigorates CD8+ T-cell immunity against glioblastoma through B-cell-dependent antigen cross-presentation in the spleen.
In Cell Mol Immunol on 1 April 2026 by Han, Y., Guo, C., et al.
PubMed
Glioblastoma multiforme (GBM) is a lethal primary brain cancer with limited treatment options. Systemic and local immunosuppression induced by GBMs contributes to malignancy aggressiveness and resistance to immune checkpoint blockade (ICB) therapy. Herein, we demonstrated that a novel oncolytic virus, M1 (OVM), reversed GBM-driven systemic immunosuppression and promoted T lymphocyte infiltration within the tumor microenvironment (TME). Intravenous administration of OVM suppressed glioma progression in a spleen-dependent manner. Mechanistically, OVM enhanced B-cell-T-cell interactions in the spleen through the formation of immune synapses. A subset of B cells positive for bone marrow stromal cell antigen 2 (Bst2) was enriched in the splenic marginal zone following OVM treatment and exhibited superior capacity for antigen cross-presentation. These splenic Bst2+ B cells activated cognate CD8+ T cells to mediate adaptive antitumor immunity against intracranial gliomas. Moreover, OVM treatment synergized with anti-PD-1 therapy and further extended the survival of glioma-bearing animals. Collectively, our findings highlight the therapeutic potential of intravenous OVM for GBM management and reveal a novel immunomodulatory mechanism underlying oncolytic virotherapy.
-
Oncolytic adenovirus delivery of neoantigens sensitizes low-mutation tumors to anti-PD-1 therapy and prevents metastasis.
In Signal Transduct Target Ther on 23 December 2025 by Shen, K. Y., Yu, S. Z., et al.
PubMed
Neoantigen vaccines and oncolytic viruses are emerging immunotherapies that can reshape the tumor microenvironment (TME). However, tumors with low mutation burdens often respond poorly to immunotherapies because of their limited immunogenicity. Developing effective immunotherapy strategies for these types of tumors remains a significant challenge. In this study, we engineered oncolytic adenoviruses to accurately amplify neoantigen expression within tumor cells, which demonstrated superior efficacy compared to synthetic long peptide vaccines and showed enhanced effectiveness in a low mutation burden intrahepatic cholangiocarcinoma model. Building on this, we further developed NeoViron, which coexpresses neoantigens and Flt3L, a dendritic cell growth factor, to promote antigen presentation and T-cell infiltration simultaneously. NeoViron significantly inhibited tumor growth and prevented metastasis in intrahepatic cholangiocarcinoma animal models. Mechanistically, NeoViron enhanced the cytotoxicity of CD8+ T cells and promoted the expansion of CD69+ CD8+ tissue-resident memory T cells and TCF1+ CD8+ stem-like T cells to promote anti-tumor immunity and immune memory. When combined with anti-PD-1, it further enhances the cytotoxicity of tissue-resident memory T cells to eradicate solid tumors. These findings demonstrate that NeoViron can effectively sensitize low-mutation tumors to immunotherapy by increasing neoantigen expression and antigen-presentation efficacy, offering a promising strategy for cancer treatment, particularly for tumors with scarce neoantigens.
-
Etoposide activates CD8+ T cell anti-tumor immunity in osteosarcoma through MHC I upregulation via tumor-secreted IL-33 mediated signaling.
In J Immunother Cancer on 21 December 2025 by He, X., Li, H., et al.
PubMed
Osteosarcoma patients with high propensity for metastasis and recurrence generally encounter a poor prognosis. Despite the extensive exploration of immunotherapy, particularly the anti-programmed cell death protein 1 (anti-PD-1) antibody, in clinical trials, the efficacy remains unsatisfactory. A more profound comprehension of the resistance mechanisms and the development of innovative therapeutic strategies is imperative.
-
Gut microbial metabolite butyrate boosts p53-expressing telomerase-specific oncolytic adenovirus efficacy by enhancing infectivity and activating MHC-I/cGAS-STING.
In Cancer Immunol Immunother on 18 December 2025 by Sakamoto, M., Kuroda, S., et al.
PubMed
The gut microbiota plays an essential role in regulating host immunity, and its metabolites such as butyrate exert immunomodulatory effects by acting as histone deacetylase inhibitors. Oncolytic virotherapy has emerged as a promising approach for cancer treatment, and we have developed OBP-702, a telomerase-specific oncolytic adenovirus that expresses p53 and elicits strong systemic antitumor responses. In this study, the potential synergy between butyrate and OBP-702 was investigated in colorectal cancer models. Using human and murine colorectal carcinoma cell lines, butyrate was found to directly enhance the infectivity of OBP-702 by upregulating CAR and integrins, thereby promoting apoptosis and autophagy in tumor cells. In addition, butyrate indirectly boosted systemic antitumor immunity by upregulating MHC-I expression through activation of the cGAS-STING pathway and enhancing CD8 + T cell recruitment via CXCL10 secretion. These findings were supported by in vivo experiments using CT26 subcutaneous, bilateral, and orthotopic tumor models, in which the combination of oral butyrate and intratumoral OBP-702 administration produced synergistic antitumor effects. These results highlight the therapeutic potential of integrating gut microbial metabolites with oncolytic virotherapy as a novel immunotherapeutic strategy for colorectal cancer.