InVivoMAb anti-mouse Ly6G/Ly6C (Gr-1)
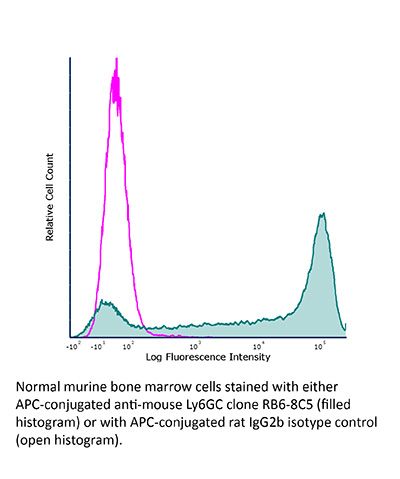
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse granulocytes |
| Reported Applications |
in vivo depletion of Gr-1+ myeloid cells Flow cytometry Immunohistochemistry (paraffin) Immunohistochemistry (frozen) |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10312146 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Schulze, F. S., et al (2014). "Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid" Am J Pathol 184(8): 2185-2196.
PubMed
Bullous pemphigoid (BP) and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases mediated by autoantibodies against type XVII collagen (Col17) and Col7, respectively. For blister formation, Fc-mediated events, such as infiltration of inflammatory cells in the skin, complement activation, and release of proteases at the dermal-epidermal junction, are essential. Although in the neonatal passive transfer mouse model of BP, tissue destruction is mediated by Fcgamma receptors (FcgammaRs) I and III, the passive transfer model of epidermolysis bullosa acquisita completely depends on FcgammaRIV. To clarify this discrepancy, we developed a novel experimental model for BP using adult mice. Lesion formation was Fc mediated because gamma-chain-deficient mice and mice treated with anti-Col17 IgG, depleted from its sugar moiety at the Fc portion, were resistant to disease induction. By the use of various FcgammaR-deficient mouse strains, tissue destruction was shown to be mediated by FcgammaRIV, FcgammaRIII, and FcgammaRIIB, whereas FcgammaRI was not essential. Furthermore, anti-inflammatory mediators in already clinically diseased mice can be explored in the novel BP model, because the pharmacological inhibition of FcgammaRIV and depletion of granulocytes abolished skin blisters. Herein, we extended our knowledge about the importance of FcgammaRs in experimental BP and established a novel BP mouse model suitable to study disease development over a longer time period and explore novel treatment strategies in a quasi-therapeutic setting.
-
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
-
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
-
Bansal, S., et al (2018). "IL-1 Signaling Prevents Alveolar Macrophage Depletion during Influenza and Streptococcus pneumoniae Coinfection" J Immunol 200(4): 1425-1433.
PubMed
Influenza and bacterial coinfection is a significant cause of hospitalization and death in humans during influenza epidemics and pandemics. However, the fundamental protective and pathogenic mechanisms involved in this complex virus-host-bacterium interaction remain incompletely understood. In this study, we have developed mild to lethal influenza and Streptococcus pneumoniae coinfection models for comparative analyses of disease pathogenesis. Specifically, wild-type and IL-1R type 1-deficient (Il1r1(-/-) ) mice were infected with influenza virus and then superchallenged with noninvasive S. pneumoniae serotype 14 (Spn14) or S. pneumoniae serotype 19A (Spn19A). The coinfections were followed by comparative analyses of inflammatory responses and animal protection. We found that resident alveolar macrophages are efficient in the clearance of both pneumococcal serotypes in the absence of influenza infection; in contrast, they are essential for airway control of Spn14 infection but not Spn19A infection. In agreement, TNF-alpha and neutrophils play a compensatory protective role in secondary bacterial infection associated with Spn19A; however, the essential requirement for alveolar macrophage-mediated clearance significantly enhances the virulence of Spn14 during postinfluenza pneumococcal infection. Furthermore, we show that, although IL-1 signaling is not required for host defense against pneumococcal infection alone, it is essential for sustaining antibacterial immunity during postinfluenza pneumococcal infection, as evidenced by significantly aggravated bacterial burden and animal mortality in Il1r1(-/-) mice. Mechanistically, we show that through preventing alveolar macrophage depletion, inflammatory cytokine IL-1 signaling is critically involved in host resistance to influenza and pneumococcal coinfection.
Product Citations
-
Neuronal Serpina3n is an endogenous protector against blood brain barrier damage following cerebral ischemic stroke.
In Journal of Cerebral Blood Flow & Metabolism on 1 February 2023 by Li, F., Zhang, Y., et al.
PubMed
Ischemic stroke results in blood-brain barrier (BBB) disruption, during which the reciprocal interaction between ischemic neurons and components of the BBB appears to play a critical role. However, the underlying mechanisms for BBB protection remain largely unknown. In this study, we found that Serpina3n, a serine protease inhibitor, was significantly upregulated in the ischemic brain, predominantly in ischemic neurons from 6 hours to 3 days after stroke. Using neuron-specific adeno-associated virus (AAV), intranasal delivery of recombinant protein, and immune-deficient Rag1-/- mice, we demonstrated that Serpina3n attenuated BBB disruption and immune cell infiltration following stroke by inhibiting the activity of granzyme B (GZMB) and neutrophil elastase (NE) secreted by T cells and neutrophils. Furthermore, we found that intranasal delivery of rSerpina3n significantly attenuated the neurologic deficits after stroke. In conclusion, Serpina3n is a novel ischemic neuron-derived proteinase inhibitor that counterbalances BBB disruption induced by peripheral T cell and neutrophil infiltration after ischemic stroke. These findings reveal a novel endogenous protective mechanism against BBB damage with Serpina3n being a potential therapeutic target in ischemic stroke.
-
Low-dose radiotherapy synergizes with PD-1 blockade to achieve durable survival in advanced NSCLC through antitumor neutrophil programming.
In Signal Transduct Target Ther on 7 May 2026 by Zhou, L., Liu, Y., et al.
PubMed
The optimal strategy for combining radiotherapy (RT) and immunotherapy remains under intensive investigation. Here we developed TRIDENT (Triple Radio-Immunotherapy-Driven ENhanced Therapy), a novel triple-modality regimen combining immunomodulatory low-dose RT (LDRT) to large tumor(s), immunogenic high-dose RT (HDRT) to small tumor(s), and PD-1 blockade. In our phase I trial of 29 patients with treatment-naïve, PD-L1-positive advanced non-small cell lung cancer (NSCLC), TRIDENT achieved a median overall survival (mOS) of 51.3 months (95% CI, 20.7-not reached), higher than outcomes typically reported with contemporary standard (chemo)immunotherapy. This durable survival signal was corroborated in an independent real-world cohort of 97 patients with advanced lung cancer (mOS: 41.5 months; 95% CI, 26.3-63.7). Mechanistically, TRIDENT elicited neutrophil-dependent, systemic antitumor immunity and induced a distinct population of antitumor TNF-α⁺ neutrophils marked by increased MHC and costimulatory molecule expression. Neutrophil recruitment was driven by the CXCL-CXCR2 axis, and polarization toward an antitumor state was programmed by treatment-induced IFN-γ and GM-CSF. TNF-α⁺ neutrophils enhanced CD8⁺ T-cell function via ICAM-1-LFA-1 interactions, and adoptive transfer confirmed their intrinsic antitumor activity in vivo. Spatial transcriptomics of patient tumor tissues further identified a TNF-α+ neutrophil-effector CD8+ T-cell niche after TRIDENT, providing a stimulatory signal to effector CD8⁺ T cells. In line with these mechanistic findings, clinical biomarker analyses linked neutrophil number with prolonged survival. TRIDENT activates an RT-driven neutrophil-CD8⁺ T-cell axis and promotes survival-associated neutrophil activation. These mechanistic insights, coupled with durable survival in our phase I trial, position TRIDENT as a promising strategy for metastatic NSCLC currently undergoing randomized phase II evaluation. Our study also highlights TNF-α+ neutrophils as a promising therapeutic strategy to enhance antitumor efficacy.
-
Hepatocyte-derived LRG1 primes the liver for metastasis and impairs immunotherapy.
In Cell Mol Immunol on 1 May 2026 by Long, G., Cheng, B., et al.
PubMed
The liver undergoes active remodeling by the primary tumor prior to metastatic spread. However, the mechanisms by which hepatocytes dictate the liver-specific tropism of tumors remain elusive. Here, we identify hepatocyte-derived leucine-rich alpha-2-glycoprotein 1 (LRG1) as a key mediator of liver premetastatic niche (PMN) formation. Clinically, elevated serum LRG1 levels are correlated with an increased risk of liver metastasis in patients and multiple mouse models. Mechanistically, LRG1 remodels the hepatic microenvironment by driving immunosuppressive neutrophil accumulation, impairing the function of effector T cells and dendritic cells, and enhancing angiogenesis in the liver, thereby fostering a prometastatic landscape. Hepatocyte-specific ablation of LRG1 dampens premetastatic niche formation and significantly reduces the metastatic burden in vivo. Hepatic LRG1 induced by tumor-associated inflammation via IL-6/STAT3 signaling promotes liver metastasis through the formation of TGFBR/PI3K/AKT axis-driven neutrophil extracellular traps (NETs). Importantly, therapeutic blockade of LRG1 not only suppressed liver metastasis but also reprogrammed the hepatic niche toward an immune-activated state, sensitizing tumors to anti-PD-1 therapy. Collectively, our findings reveal a hepatocyte-LRG1 axis that drives liver premetastatic niche remodeling and highlight LRG1 as a promising target for the prevention and treatment of liver metastasis.
-
Low-dose oral nicotinamide mononucleotide for immune thrombocytopenia: a phase 1/2 trial.
In Nat Med on 29 April 2026 by Li, H., Xu, Y., et al.
PubMed
Autoimmunity remains challenging to treat without broad immunosuppression. We previously showed that anti-CD38 antibody can rapidly elevate platelet counts in refractory immune thrombocytopenia (ITP), but the underlying mechanism was unclear. Here we report that anti-CD38 antibody induces platelet recovery within 3 days, including after retreatment in relapsed cases. Mechanistically, CD38-mediated nicotinamide adenine dinucleotide (NAD+) depletion drives M1-like macrophage polarization with increased Fc gamma receptor I (FcγRI) expression, thereby promoting macrophage phagocytosis of opsonized platelets. In mice, CD38 inhibition or nicotinamide mononucleotide (NMN) supplementation restores NAD+, reprograms macrophages, downregulates FcγRI and prevents thrombocytopenia. In an ovalbumin immunization model, NMN treatment does not impair antigen-specific antibody production, supporting preservation of humoral responses. Based on these findings, we conducted a single-arm, open-label phase 1/2 trial of low-dose oral NMN (450 mg twice daily for 2 weeks) in adults with steroid-refractory or steroid-dependent ITP. Primary endpoints were safety/tolerability and platelet response (≥50 × 109 per liter within 2 weeks, confirmed by two consecutive measurements one or more days apart, without rescue therapy or dose escalation of thrombopoietin receptor agonists or corticosteroids). Among 25 enrolled patients, no dose-limiting toxicities or treatment-related serious adverse events occurred; NMN was well tolerated, with only mild treatment-related adverse events in 12% and non-severe infections (grade 1) in 8% of patients while immunoglobulin levels remained stable, consistent with preserved humoral immunity. Five patients (20.0%) met the primary platelet-response endpoint. In exploratory analyses, overall, 60% of patients achieved platelet counts more than 1.5× baseline during treatment, and 52% maintained responses through week 8. Together, these data identify the CD38-NAD+ axis as an immunometabolic checkpoint in ITP and support further exploration of NMN as a non-antibody-depleting metabolic strategy for antibody-mediated disease. ClinicalTrials.gov identifier: NCT06776510 .