InVivoMAb anti-mouse IL-10R (CD210)
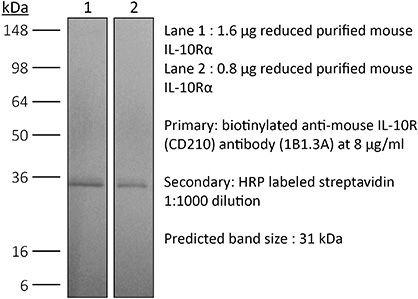
Product Description
Bio X Cell is pleased to offer two recombinant, murine chimeric versions of the original 1B1.3A antibody, 1B1.3A-CP060 and 1B1.3A-CP073. The variable region sequences are identical to the original 1B1.3A but the constant region sequences have been switched from Rat IgG1, κ to mouse IgG2a, κ for use in murine models. Species-matched chimeric antibodies exhibit regulated effector functions—including Fc receptor binding and complement activation—and cause less immunogenicity and formation of anti-drug antibodies (ADAs) than xenogenic antibodies in animal models. Additionally, 1B1.3A-CP073 contains LALA-PG Fc-silencing mutations in the heavy chain rendering it unable to bind endogenous murine FcγR or C1q to induce antibody-dependent, cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). Antibodies with active Fc regions can engage immune cells via FcγRs, leading to the depletion of antigen expressing cells through mechanisms like ADCC or complement activation. Fc-silenced antibodies do not trigger these pathways and can block signaling without killing or depleting target cells. The highly controlled sequence and lack of genetic drift in recombinant antibodies provide more reliable and reproducible results over hybridoma derived antibodies.
Specifications
| Isotype | Rat IgG1, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG1 isotype control, anti-horseradish peroxidase |
| Recommended Dilution Buffer | InVivoPure pH 6.5T Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant ligand-binding domain of mouse IL-10R |
| Reported Applications |
in vivo blocking of IL-10/IL-10R signaling in vitro blocking of IL-10R signaling Flow cytometry Western Blot |
| Formulation |
PBS, pH 6.5 0.01% Tween Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107611 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Hu, Z., et al (2013). "Regulatory CD8+ T cells associated with erosion of immune surveillance in persistent virus infection suppress in vitro and have a reversible proliferative defect" J Immunol 191(1): 312-322.
PubMed
CD4(+) T cell help is critical for CD8(+) T cell memory and immune surveillance against persistent virus infections. Our recent data have showed the lack of CD4(+) T cells leads to the generation of an IL-10-producing CD8(+) T cell population during persistent murine gamma-herpesvirus-68 (MHV-68) infection. IL-10 from these cells is partly responsible for erosion in immune surveillance, leading to spontaneous virus reactivation in lungs. In this study, we further characterized the generation, phenotype, and function of these IL-10-producing CD8(+) T cells by comparing with a newly identified IL-10-producing CD8(+) T cell population present during the acute stage of the infection. The IL-10-producing CD8(+) populations in acute and chronic stages differed in their requirement for CD4(+) T cell help, the dependence on IL-2/CD25 and CD40-CD40L pathways, and the ability to proliferate in vitro in response to anti-CD3 stimulation. IL-10-producing CD8(+) T cells in the chronic stage showed a distinct immunophenotypic profile, sharing partial overlap with the markers of previously reported regulatory CD8(+) T cells, and suppressed the proliferation of naive CD8(+) T cells. Notably, they retained the ability to produce effector cytokines and cytotoxic activity. In addition, the proliferative defect of the cells could be restored by addition of exogenous IL-2 or blockade of IL-10. These data suggest that the IL-10-producing CD8(+) T cells arising in chronic MHV-68 infection in the absence of CD4(+) T cell help belong to a subset of CD8(+) regulatory T cells.
-
Christensen, A. D., et al (2015). "Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells" Clin Exp Immunol 179(3): 485-499.
PubMed
Regulatory T cells (Tregs ) are known to play an immunosuppressive role in the response of contact hypersensitivity (CHS), but neither the dynamics of Tregs during the CHS response nor the exaggerated inflammatory response after depletion of Tregs has been characterized in detail. In this study we show that the number of Tregs in the challenged tissue peak at the same time as the ear-swelling reaches its maximum on day 1 after challenge, whereas the number of Tregs in the draining lymph nodes peaks at day 2. As expected, depletion of Tregs by injection of a monoclonal antibody to CD25 prior to sensitization led to a prolonged and sustained inflammatory response which was dependent upon CD8 T cells, and co-stimulatory blockade with cytotoxic T lymphocyte antigen-4-immunoglobulin (CTLA-4-Ig) suppressed the exaggerated inflammation. In contrast, blockade of the interleukin (IL)-10-receptor (IL-10R) did not further increase the exaggerated inflammatory response in the Treg -depleted mice. In the absence of Tregs , the response changed from a mainly acute reaction with heavy infiltration of neutrophils to a sustained response with more chronic characteristics (fewer neutrophils and dominated by macrophages). Furthermore, depletion of Tregs enhanced the release of cytokines and chemokines locally in the inflamed ear and augmented serum levels of the systemic inflammatory mediators serum amyloid (SAP) and haptoglobin early in the response.
-
Dolina, J. S., et al (2014). "Liver-primed CD8+ T cells suppress antiviral adaptive immunity through galectin-9-independent T-cell immunoglobulin and mucin 3 engagement of high-mobility group box 1 in mice" Hepatology 59(4): 1351-1365.
PubMed
The liver is a tolerogenic environment exploited by persistent infections, such as hepatitis B (HBV) and C (HCV) viruses. In a murine model of intravenous hepatotropic adenovirus infection, liver-primed antiviral CD8(+) T cells fail to produce proinflammatory cytokines and do not display cytolytic activity characteristic of effector CD8(+) T cells generated by infection at an extrahepatic, that is, subcutaneous, site. Importantly, liver-generated CD8(+) T cells also appear to have a T-regulatory (Treg) cell function exemplified by their ability to limit proliferation of antigen-specific T-effector (Teff ) cells in vitro and in vivo via T-cell immunoglobulin and mucin 3 (Tim-3) expressed by the CD8(+) Treg cells. Regulatory activity did not require recognition of the canonical Tim-3 ligand, galectin-9, but was dependent on CD8(+) Treg cell-surface Tim-3 binding to the alarmin, high-mobility group box 1 (HMGB-1). CONCLUSION: Virus-specific Tim-3(+) CD8(+) T cells operating through HMGB-1 recognition in the setting of acute and chronic viral infections of the liver may act to dampen hepatic T-cell responses in the liver microenvironment and, as a consequence, limit immune-mediated tissue injury or promote the establishment of persistent infections.
-
Burrack, K. S., et al (2018). "Interleukin-15 Complex Treatment Protects Mice from Cerebral Malaria by Inducing Interleukin-10-Producing Natural Killer Cells" Immunity 48(4): 760-772 e764.
PubMed
Cerebral malaria is a deadly complication of Plasmodium infection and involves blood brain barrier (BBB) disruption following infiltration of white blood cells. During experimental cerebral malaria (ECM), mice inoculated with Plasmodium berghei ANKA-infected red blood cells develop a fatal CM-like disease caused by CD8(+) T cell-mediated pathology. We found that treatment with interleukin-15 complex (IL-15C) prevented ECM, whereas IL-2C treatment had no effect. IL-15C-expanded natural killer (NK) cells were necessary and sufficient for protection against ECM. IL-15C treatment also decreased CD8(+) T cell activation in the brain and prevented BBB breakdown without influencing parasite load. IL-15C induced NK cells to express IL-10, which was required for IL-15C-mediated protection against ECM. Finally, we show that ALT-803, a modified human IL-15C, mediates similar induction of IL-10 in NK cells and protection against ECM. These data identify a regulatory role for cytokine-stimulated NK cells in the prevention of a pathogenic immune response.
Product Citations
-
Antigen choice determines vaccine-induced generation of immunogenic versus tolerogenic dendritic cells that are marked by differential expression of pancreatic enzymes.
In The Journal of Immunology on 1 April 2013 by Farkas, A. M., Marvel, D. M., et al.
PubMed
Dendritic cells (DC) elicit immunity to pathogens and tumors while simultaneously preserving tolerance to self. Efficacious cancer vaccines have been a challenge because they are based on tumor Ags, some of which are self-Ags and thus subject to self-tolerance. One such Ag is the tumor-associated mucin MUC1. Preclinical testing of MUC1 vaccines revealed existence of peripheral tolerance to MUC1 that compromises their efficacy. To identify mechanisms that act early postvaccination and might predict vaccine outcome, we immunized human MUC1 transgenic mice (MUC1.Tg) i.v. with a MUC1 peptide vaccine against which they generate weak immunity and wild-type (WT) mice that respond strongly to the same peptide. We analyzed differences in splenic DC phenotype and function between the two mouse strains at 24 and 72 h postvaccination and also performed unbiased total gene expression analysis of the spleen. Compared to WT, MUC1.Tg spleens had significantly fewer DC, and they exhibited significantly lower expression of costimulatory molecules, decreased motility, and preferential priming of Ag-specific Foxp3(+) regulatory T cells. This tolerogenic DC phenotype and function was marked by a new putative biomarker revealed by the microarray: a cohort of pancreatic enzymes (trypsin, carboxypeptidase, elastase, and others) not previously reported in DC. These enzymes were strongly upregulated in the splenic DC from vaccinated WT mice and suppressed in the splenic DC of vaccinated MUC1.Tg mice. Suppression of the enzymes was dependent on regulatory T cells and on signaling through the IL-10R and correlated with global downregulation of DC immunostimulatory phenotype and function.
-
Identification of distinct cDC2 subpopulations that direct microbiota-specific T cell differentiation
In bioRxiv on 5 November 2025 by Carroll, S. L., Ly, A., et al.
-
TCF1 and LEF1 promote B-1a cell homeostasis and regulatory function.
In Nature on 1 October 2025 by Shen, Q., Wang, H., et al.
PubMed
B-1 cells are innate-like immune cells abundant in serosal cavities with antibodies enriched in bacterial recognition, yet their existence in humans has been controversial1-3. The CD5+ B-1a subset expresses anti-inflammatory molecules including IL-10, PDL1 and CTLA4 and can be immunoregulatory4-6. Unlike conventional B cells that are continuously replenished, B-1a cells are produced early in life and maintained through self-renewal7. Here we show that the transcription factors TCF1 and LEF1 are critical regulators of B-1a cells. LEF1 expression is highest in fetal and bone marrow B-1 progenitors, whereas the levels of TCF1 are higher in splenic and peritoneal B-1 cells than in B-1 progenitors. TCF1-LEF1 double deficient mice have reduced B-1a cells and defective B-1a cell maintenance. These transcription factors promote MYC-dependent metabolic pathways and induce a stem-like population upon activation, partly via IL-10 production. In the absence of TCF1 and LEF1, B-1 cells proliferate excessively and acquire an exhausted phenotype with reduced IL-10 and PDL1 expression. Furthermore, adoptive transfer of B-1 cells lacking TCF1 and LEF1 fails to suppress brain inflammation. These transcription factors are also expressed in human chronic lymphocytic leukaemia B cells and in a B-1-like population that is abundant in pleural fluid and circulation of some patients with pleural infection. Our findings define a TCF1-LEF1-driven transcriptional program that integrates stemness and regulatory function in B-1a cells.
-
Interleukin-10 limits immune-mediated pathology in chronic subclinical plasmodial infection.
In PLoS Negl Trop Dis on 1 September 2025 by Silva, L. S., Monks, B. G., et al.
PubMed
Subclinical parasitemia constitutes the predominant proportion of Plasmodium spp. infections in hyperendemic regions of the world. Elevated levels of serum interleukin-10 (IL-10) are observed in both acute symptomatic and chronic subclinical Plasmodium spp. infections. The role of IL-10 in acute infection has been extensively studied; however, the role of sustained elevated levels of IL-10 in chronic subclinical plasmodial infections remains to be determined. We investigated the role of IL-10 in a long-term subclinical and patent Plasmodium chabaudi chabaudi-AS (Pc) infection using mice lacking humoral immunity (µMT-/- mice). Pc-infected µMT-/- mice exhibit a long-term (99 days) chronic infection, with microscopic levels of parasitemia and without any outward signs of disease. We found that chronically infected mice have slightly elevated levels of tumor necrosis factor α (TNFα) and interferon-γ (IFNγ), and high levels of IL-10 in the circulation. The source of IL-10 was CD4+ T cells. We found that elevated IL-10 levels were mechanistically linked to subclinical Plasmodium infection by blocking IL-10 signaling. Anti-IL-10R resulted in a marked, albeit transient, reduction of the parasitemia that was accompanied by a robust pro-inflammatory response and death of chronically infected µMT-/- mice. A similar outcome was observed in infected µMT-/- mice after CD4+ T cell depletion with anti-CD4 antibody. CD4-depleted infected µMT-/- mice exhibited reduced IL-10 and rapid weight loss, succumbing to infection by day 6 after CD4 neutralization. Our results showed that IL-10 from CD4+ T cells limits immune-mediated pathology in chronic subclinical Pc infection in µMT-/- mice by protecting against excessive inflammatory responses to blood-stage parasites.