InVivoMAb anti-mouse Thy1.2 (CD90.2)
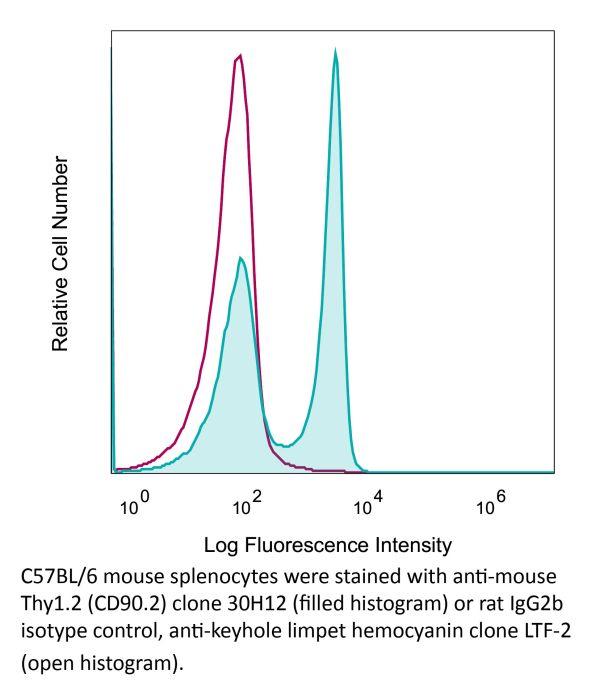
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse thymus or spleen |
| Reported Applications |
in vivo ILC depletion in vivo T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107682 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
-
Gladiator, A., et al (2013). "Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection" J Immunol 190(2): 521-525.
PubMed
IL-17-mediated immunity has emerged as a crucial host defense mechanism against fungal infections. Although Th cells are generally thought to act as the major source of IL-17 in response to Candida albicans, we show that fungal control is mediated by IL-17-secreting innate lymphoid cells (ILCs) and not by Th17 cells. By using a mouse model of oropharyngeal candidiasis we found that IL-17A and IL-17F, which are both crucial for pathogen clearance, are produced promptly upon infection in an IL-23-dependent manner, and that ILCs in the oral mucosa are the main source for these cytokines. Ab-mediated depletion of ILCs in RAG1-deficient mice or ILC deficiency in retinoic acid-related orphan receptor c(-/-) mice resulted in a complete failure to control the infection. Taken together, our data uncover the cellular basis for the IL-23/IL-17 axis, which acts right at the onset of infection when it is most needed for fungal control and host protection.
-
Brasseit, J., et al (2015). "CD4 T cells are required for both development and maintenance of disease in a new mouse model of reversible colitis" Mucosal Immunol. doi : 10.1038/mi.2015.93.
PubMed
Current therapies to treat inflammatory bowel diseases have limited efficacy, significant side effects, and often wane over time. Little is known about the cellular and molecular mechanisms operative in the process of mucosal healing from colitis. To study such events, we developed a new model of reversible colitis in which adoptive transfer of CD4+CD45RBhi T cells into Helicobacter typhlonius-colonized lymphopenic mice resulted in a rapid onset of colonic inflammation that was reversible through depletion of colitogenic T cells. Remission was associated with an improved clinical and histopathological score, reduced immune cell infiltration to the intestinal mucosa, altered intestinal gene expression profiles, regeneration of the colonic mucus layer, and the restoration of epithelial barrier integrity. Notably, colitogenic T cells were not only critical for induction of colitis but also for maintenance of disease. Depletion of colitogenic T cells resulted in a rapid drop in tumor necrosis factor alpha (TNFalpha) levels associated with reduced infiltration of inflammatory immune cells to sites of inflammation. Although neutralization of TNFalpha prevented the onset of colitis, anti-TNFalpha treatment of mice with established disease failed to resolve colonic inflammation. Collectively, this new model of reversible colitis provides an important research tool to study the dynamics of mucosal healing in chronic intestinal remitting-relapsing disorders.
-
Monticelli, L. A., et al (2011). "Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus" Nat Immunol 12(11): 1045-1054.
PubMed
Innate lymphoid cells (ILCs), a heterogeneous cell population, are critical in orchestrating immunity and inflammation in the intestine, but whether ILCs influence immune responses or tissue homeostasis at other mucosal sites remains poorly characterized. Here we identify a population of lung-resident ILCs in mice and humans that expressed the alloantigen Thy-1 (CD90), interleukin 2 (IL-2) receptor a-chain (CD25), IL-7 receptor a-chain (CD127) and the IL-33 receptor subunit T1-ST2. Notably, mouse ILCs accumulated in the lung after infection with influenza virus, and depletion of ILCs resulted in loss of airway epithelial integrity, diminished lung function and impaired airway remodeling. These defects were restored by administration of the lung ILC product amphiregulin. Collectively, our results demonstrate a critical role for lung ILCs in restoring airway epithelial integrity and tissue homeostasis after infection with influenza virus.
Product Citations
-
Nerves Stimulate Cross-talk Between Gastric Cancer and Group 3 Innate Lymphoid Cells to Enhance Immunosuppression.
In Cancer Res on 15 April 2026 by Liao, F., Tong, Y., et al.
PubMed
The immunosuppressive tumor microenvironment (TME) enables cancer cells to evade clinical immunotherapies. Neural networks are vital components of the TME, and interactions among cancer cells, neuronal cells, and immune cells mediate immunosuppression. Hence, understanding the mechanisms of intercellular cross-talk could inform immunomodulatory approaches to enhance immunotherapy efficacy. In this study, we found that the vagus nerve regulated the cross-talk between gastric cancer cells and group 3 innate lymphoid cells (ILC3), boosting immune resistance in gastric cancer by enhancing programmed cell death ligand 1 (PD-L1) expression. Specifically, the infiltrated vagus nerve released acetylcholine (ACh) that elevated the expression of lipase ABHD16A in gastric cancer cells, facilitating the production and secretion of the metabolite lysophosphatidylserine (LysoPS) into the TME. LysoPS facilitated the proliferation and activation of ILC3s in the TME, resulting in the production of the cytokine IL22 via the GPR34-AKT-STAT3 axis. In turn, IL22 triggered the unfolded protein response (UPR) in gastric cancer cells, which led to an increase in PD-L1 expression that enhanced immune resistance. Importantly, targeting ACh or the cross-talk between gastric cancer cells and ILC3s significantly enhanced the efficacy of anti-PD-L1 immunotherapy. Serum levels of LysoPS and IL22 were elevated in patients with gastric cancer, particularly those with perineural invasion. Collectively, these findings provide valuable insights into the cross-talk among gastric cancer cells, nerve cells, and ILC3s that regulate immunosuppression and response to anti-PD-L1 immunotherapy, emphasizing the potential clinical significance of this axis for detecting and treating gastric cancer.
-
Nerves Stimulate Cross-talk Between Gastric Cancer and Group 3 Innate Lymphoid Cells to Enhance Immunosuppression.
In Cancer Res on 15 April 2026 by Liao, F., Tong, Y., et al.
PubMed
The immunosuppressive tumor microenvironment (TME) enables cancer cells to evade clinical immunotherapies. Neural networks are vital components of the TME, and interactions among cancer cells, neuronal cells, and immune cells mediate immunosuppression. Hence, understanding the mechanisms of intercellular cross-talk could inform immunomodulatory approaches to enhance immunotherapy efficacy. In this study, we found that the vagus nerve regulated the cross-talk between gastric cancer cells and group 3 innate lymphoid cells (ILC3), boosting immune resistance in gastric cancer by enhancing programmed cell death ligand 1 (PD-L1) expression. Specifically, the infiltrated vagus nerve released acetylcholine (ACh) that elevated the expression of lipase ABHD16A in gastric cancer cells, facilitating the production and secretion of the metabolite lysophosphatidylserine (LysoPS) into the TME. LysoPS facilitated the proliferation and activation of ILC3s in the TME, resulting in the production of the cytokine IL22 via the GPR34-AKT-STAT3 axis. In turn, IL22 triggered the unfolded protein response (UPR) in gastric cancer cells, which led to an increase in PD-L1 expression that enhanced immune resistance. Importantly, targeting ACh or the cross-talk between gastric cancer cells and ILC3s significantly enhanced the efficacy of anti-PD-L1 immunotherapy. Serum levels of LysoPS and IL22 were elevated in patients with gastric cancer, particularly those with perineural invasion. Collectively, these findings provide valuable insights into the cross-talk among gastric cancer cells, nerve cells, and ILC3s that regulate immunosuppression and response to anti-PD-L1 immunotherapy, emphasizing the potential clinical significance of this axis for detecting and treating gastric cancer.
-
Long-term inhibition of protease hypersensitivity by initial immunological cross-regulation and epigenetic memory in lung stromal cells.
In Nat Immunol on 1 April 2026 by Ryu, J., Blondeau, A., et al.
PubMed
Prevention and regulation of excessive inflammation is a key target to protect against inflammatory pathologies such as autoimmunity and allergy. In a mouse model of acute lung protease hypersensitivity, we assessed the efficacy of immunological cross-regulation to mitigate pathogenic inflammation. We show that induction of a type 1 response using Toll-like receptor ligands or a bacterial lysate efficiently blocks acute eosinophilia and type 2 responses evoked by the cysteine protease papain. Upon rechallenge with papain weeks later, mice displayed enhanced type 2 responses and eosinophilia, whereas this response was absent if the initial inflammation was cross-regulated. Memory of the initial event was stored in adventitial stromal cells expressing CCL11. Accessibility of the Ccl11 locus was increased by papain exposure in an interleukin-4- and interleukin-13-dependent manner and blocked by interferon gamma. Our results show how the nature of an initial inflammation is memorized by tissue-resident cells and shapes subsequent inflammatory responses.
-
Lung tissue-resident memory T cells optimize protection by IL-10 regulation of innate immunity.
In J Exp Med on 5 January 2026 by Yang, A. Y., Davis-Porada, J., et al.
PubMed
Respiratory viral infections establish tissue-resident memory T cells (TRM) in the lung, which provide optimal protection against subsequent infections, though the underlying mechanisms are incompletely understood. Here, we demonstrate in a mouse model of heterosubtypic influenza infection that lung TRM attenuate inflammation by macrophages during secondary versus primary responses, in part, through production of the immunoregulatory cytokine IL-10. During secondary infections, lung TRM were the predominant producers of early IL-10; inhibiting early IL-10 signaling resulted in increased macrophage-mediated inflammation, morbidity, and lung pathology. Moreover, lung TRM were shown to directly modulate lung macrophage responses and polarization in depletion experiments. Finally, IL-10 enhanced IFN-γ production by lung memory CD8+ T cells. Human influenza-specific TRM isolated from lungs recapitulated robust IL-10 expression associated with augmented effector responses of murine TRM. These data support a dual role of TRM in coordinating in situ secondary responses-augmenting effector responses for robust viral clearance while dampening inflammation to limit tissue damage.